




La paraparesia espástica hereditaria es un grupo de trastornos neurodegenerativos con heterogeneidad fenotípica y genética, caracterizados clínicamente por espasticidad y debilidad progresiva de los miembros inferiores, y en los que se describen formas de herencia autosómica dominante, autosómica recesiva y ligada al X. Actualmente hay 72 tipos descritos, de los cuales no todos tienen una mutación causal claramente definida.
Presentamos el caso de un varón de 7 años con paraparesia espástica familiar MRD9 en relación con la variante patogénica en heterocigosis en el gen Kinesin Family Member 1A (KIF1A) (mutación tipo missence en Chr 2:241724479C/T; exón 7, con modificación de p.Arg216His).
El paciente acude por primera vez a consulta traído por sus progenitores a los 3 años de edad por percibir cierto retraso psicomotor: sostén cefálico a los 2,5 meses, sedestación a los 10 meses, primeros bisílabos con 13-16 meses y marcha a los 21 meses. El niño había comenzado la escolarización, pero mostraba mayor lentitud de aprendizaje con respecto a sus compañeros, con un lenguaje más pobre (si bien la comprensión estaba conservada). A nivel de antecedentes, el embarazo cursó con normalidad, realizándose una cesárea a las 38 semanas, con un APGAR 10/10, siendo el periodo neonatal completamente normal (pruebas endocrino metabólicas y desarrollo ponderoestatural normales). No existía consanguinidad familiar.
A la exploración destacaba hiperreflexia en miembros inferiores (especialmente en hemicuerpo derecho), con un reflejo cutáneo plantar extensor bilateral y una marcha espástica. No se constataron alteraciones en la fuerza motora (mínimo aumento de tono en miembro inferior derecho) ni en la sensibilidad, tampoco dismetrías.
Fue además diagnosticado de vejiga neurógena. El paciente fue también valorado por parte de oftalmología y otorrinolaringología, sin otros hallazgos aparte de astigmatismo en ambos ojos.
Ante la sospecha de paraparesia espástica de posible causa genética vs. secundaria a parálisis cerebral infantil se realiza una RM cráneo-medular, así como un estudio de X-frágil y un cariotipo que son normales. Los ácidos pristánico, fitánico y grasos de cadena muy larga fueron también normales. Analíticamente: CK de 189, cobalamina 603pg/ml, vitamina D 19ng/ml, vitamina E 12,2μg/ml, anticuerpos contra la gliadina deaminada y contra la transglutaminasa negativos.
Posteriormente se solicitan potenciales evocados somestésicos, auditivos y visuales que son normales (apenas un discreto incremento bilateral de latencia de P100 probablemente relacionado con defecto de refracción), así como un electroneurograma que muestra una polineuropatía sensitiva axonal de intensidad moderada. En la RM craneal de control realizada se visualiza un discreto grado de atrofia cerebelosa (vermis y hemisferios) (fig. 1) con ampliación de espacios cisternales, no detectada en la RM previa 3 años atrás.
; visualización de atrofia cerebelosa moderada.")
Ante estos resultados, se solicita secuenciación del exoma humano para identificar variantes genómicas en 122 genes asociados a paraparesia espástica y ataxias, detectándose la variante patogénica en heterocigosis en el gen KIF1A. Se realiza estudio a los padres que no son portadores de la mutación.
Evolutivamente se ha ido observando una mejoría de la marcha, así como de la motricidad fina si bien padece de déficit de atención y de problemas para la escritura fina. El paciente ha referido ocasionalmente molestias en los dedos de las manos en relación con la actividad física. Desde los 6 años es capaz de controlar esfínteres, manteniendo seguimiento por parte de nefrología.
A día de hoy, el paciente sigue escolarizado y aunque precisa de mucho apoyo, es capaz de leer y de escribir. A reseñar que tras introducción de metilfenidato se constata notable mejoría del rendimiento académico.
El panel utilizado para la preparación de la librería ha sido diseñado mediante tecnología Ion AmpliSeq™ Exome (Life Technologies), captura >97% de las CCDS (>19.000 genes, >198.000 exones, >85% de las alteraciones responsables de las enfermedades de origen genético) y de las regiones de splicing flanqueantes (5bp), tiene un tamaño de ∼33Mb y comprende un total de 293.903 amplicones. La secuenciación de la librería se realizó en un secuenciador masivo de última generación, Ion Proton™ (Life Technologies). Las secuencias obtenidas han sido alineadas frente al genoma de referencia (build 37 del genoma Hg19), con el software TMAP –Ion-Alignment. Las secuencias alineadas y filtradas, según criterios de calidad específicos, fueron analizadas para la identificación de las variaciones de los nucleótidos respecto al genoma de referencia, variantes(variant-Caller). La anotación de las variantes se ha realizado con la última versión disponible de Ion Reporter™ (Life Technologies), que permite, en el caso del análisis de tríos, la anotación de las variantes del probando según el tipo de categoría genética. El análisis realizado está dirigido a la identificación de variantes incluidas en regiones exónicas o regiones de splicing, que impliquen una modificación a nivel de proteína (mutaciones de cambio de sentido o sin sentido e inserciones, deleciones o indels de nucleótidos), que se encuentren en una frecuencia superior al 40% de las lecturas. La lista de las variantes identificadas ha sido evaluada con la información de la base de datos de polimorfismos descritos (http://www.ncbi.nlm.nih.gov/SNP/, http://www.1000genomes.org, y http://evs.gs.washington.edu/EVS) para identificar variantes benignas que suelen existir en la población general y que no han sido asociadas a enfermedades (variantes presentes al menos en un 1% de la población).
Asimismo, se ha estimado el efecto funcional de las variaciones genómicas clasificadas como patogénicas utilizando 7 sistemas de predicción (SIFT, PROVEAN, PolyPhen2, MutationTaster, MutationAssessor, LRT y FATHMM) incluidos en el paquete de análisis de ALAMUT (http://www.interactivebiosoftware.com) y en el paquete ANNOVAR (http://www.openbioinformatics.org/annovar/). Finalmente, se ha evaluado la asociación de las mutaciones identificadas con los síndromes OMIM.
La variante c.647G>A en el gen KIF1A ha sido identificada mediante secuenciación masiva del exoma humano con el estudio de 122 genes asociados a la paraparesia espástica y a ataxias. El número medio de lecturas para el estudio realizado ha sido de 90. La profundidad de lectura de los amplicones incluidos en el estudio ha sido inferior a 10X en el 4,4% de los amplicones secuenciados.
La variante en el gen KIF1A ha sido confirmada mediante secuenciación directa por el método de Sanger. Dicha variante está presente en heterocigosis en la muestra del paciente.
La variante KIF1A c.647G>A (p.Arg216His), se trata de una variante de cambio de sentido que afecta al dominio motor de la proteína y que ha sido registrada como una variante patogénica en la base de datos HGMD (accession: CM157166). Consiste en el cambio de nucleótido c.647G>A (NM_001244008.1) en el exón 7 del gen KIF1Ade 49 exones y localizado en la citobanda 2q37.3. Es por tanto una variante exónica (exón 7 del gen KIF1A). Este cambio da lugar a la sustitución de una arginina por una histidina en la posición 216 de la proteína (p.Arg216His), generando una mutación de cambio de sentido.
Esta variante ha sido registrada en la base de datos dbSNP con el identificador rs672601368, sin frecuencia alélica asociada. Los resultados de los sistemas de análisis bioinformático de predicción del efecto de las mutaciones indican, en 6 de los 7 predictores utilizados (PROVEAN, SIFT, PolyPhen2, MutationTaster,MutationAssessor y FATHMM), que se trata de una variante deletérea4-9.
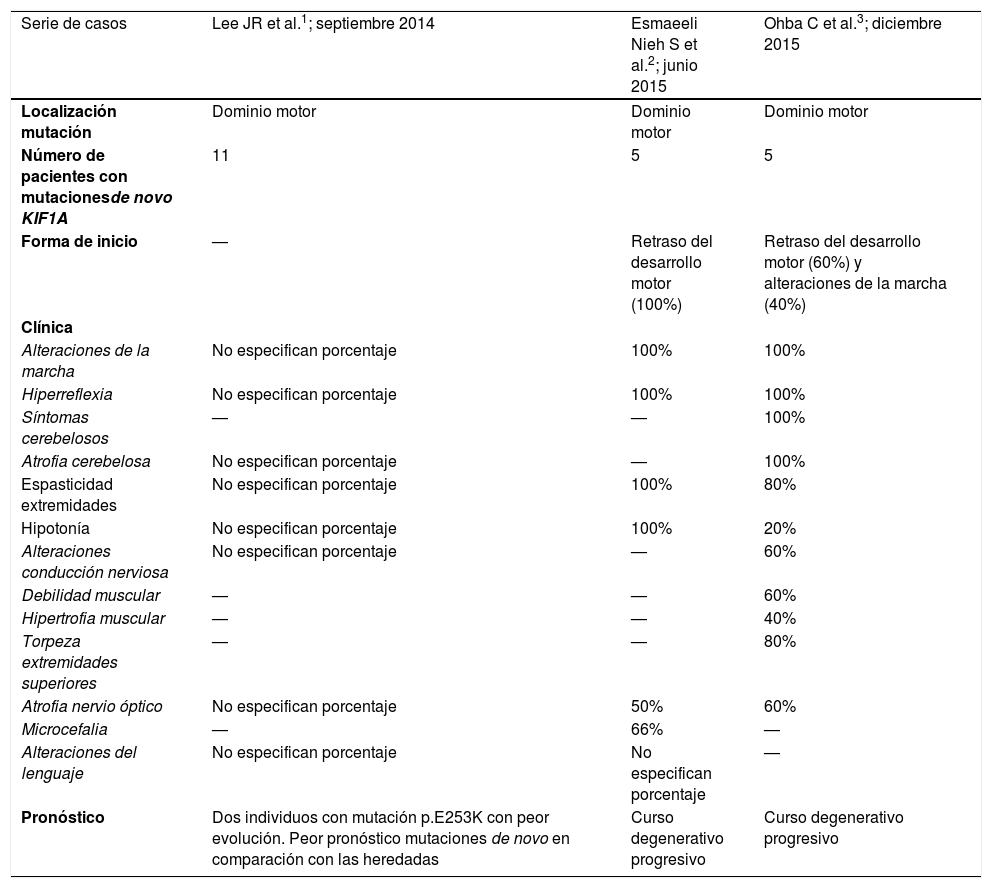
La variante ha sido descrita previamente en un paciente varón de 16 años que presenta neuropatía periférica, paraparesia espástica y retraso motor y del lenguaje, entre otras características clínicas; y en el que se identificaron 2 variantes de novo: la variante KIF1A c.647G>A y una variante en NID1. En este paciente se postula que la clínica que presenta se debe principalmente a la variante identificada en KIF1A. La presencia de variantes de cambio de sentido denovo en el dominio motor de KIF1A se ha asociado a un cuadro clínico caracterizado por discapacidad intelectual, paraparesia espástica, neuropatía axonal y atrofia cerebelar que generalmente llevan a un deterioro progresivo con mala evolución (tabla 1)10-13.
Recapitulación de las características clínicas y genéticas de los pacientes con mutaciones de novo del gen KIF1A obtenidas de las series de casos publicadas en la literatura
| Serie de casos | Lee JR et al.1; septiembre 2014 | Esmaeeli Nieh S et al.2; junio 2015 | Ohba C et al.3; diciembre 2015 |
| Localización mutación | Dominio motor | Dominio motor | Dominio motor |
| Número de pacientes con mutacionesde novo KIF1A | 11 | 5 | 5 |
| Forma de inicio | — | Retraso del desarrollo motor (100%) | Retraso del desarrollo motor (60%) y alteraciones de la marcha (40%) |
| Clínica | |||
| Alteraciones de la marcha | No especifican porcentaje | 100% | 100% |
| Hiperreflexia | No especifican porcentaje | 100% | 100% |
| Síntomas cerebelosos | — | — | 100% |
| Atrofia cerebelosa | No especifican porcentaje | — | 100% |
| Espasticidad extremidades | No especifican porcentaje | 100% | 80% |
| Hipotonía | No especifican porcentaje | 100% | 20% |
| Alteraciones conducción nerviosa | No especifican porcentaje | — | 60% |
| Debilidad muscular | — | — | 60% |
| Hipertrofia muscular | — | — | 40% |
| Torpeza extremidades superiores | — | — | 80% |
| Atrofia nervio óptico | No especifican porcentaje | 50% | 60% |
| Microcefalia | — | 66% | — |
| Alteraciones del lenguaje | No especifican porcentaje | No especifican porcentaje | — |
| Pronóstico | Dos individuos con mutación p.E253K con peor evolución. Peor pronóstico mutaciones de novo en comparación con las heredadas | Curso degenerativo progresivo | Curso degenerativo progresivo |
El caso que presentamos, sin embargo muestra una cierta estabilidad clínica presentando una mejoría en varias áreas tanto físicas como cognitivas.
El uso del exoma de forma precoz podría suponer un ahorro material y temporal a la hora de conseguir un diagnóstico.

