



Los espasmos tónicos paroxísticos dolorosos (ETPD) fueron descriptos inicialmente en la esclerosis múltiple (EM) pero serían más frecuentes en la neuromielitis óptica (NMO). El objetivo es comunicar su presencia en una serie de casos de NMO y su espectro (NMOSD), determinar la frecuencia y las características clínicas.
Métodos y pacientesSe evaluaron retrospectivamente historias clínicas de pacientes con NMO/NMOSD en 2 centros de la Ciudad de Buenos Aires (Hospital Durand y Hospital Álvarez) durante el periodo 2009-2013.
ResultadosDe 15 pacientes con NMOSD (7 con NMO definida y 8 con NMO limitada), 4 presentaron ETPD (26,66%). En los pacientes con NMO definida la frecuencia fue del 57,14% (4/7). De 9 (9/15) pacientes con mielitis longitudinal extensa (LETM) 44,44% presentó ETPD. Edad: media 35 años (rango: 22-38 años). Cien por cien sexo femenino. Tiempo desde el diagnóstico de NMO: media 7 meses (rango: 1-29 meses) y con respecto a la última recaída de LETM: media 30 días (rango: 23-40 días). El 100% presentó LETM (cervicodorsal 75% y dorsal 25%) en resonancia magnética (RM). El 100% presentó control de los espasmos y el dolor con carbamazepina (uno asociado a gabapentin) sin una respuesta adecuada a pregabalina, gabapentin y fenitoína.
ConclusionesLos ETPD son frecuentes en la NMO. Aparecen aproximadamente al mes de una recaída de LETM con lesiones cervicodorsales extensas en RM. Tienen excelente respuesta a carbamazepina y poca o nula a pregabalina y gabapentin. Estos resultados deberán ser confirmados con estudios prospectivos con mayor número de pacientes.
Paroxysmal painful tonic spasms (PPTS) were initially described in multiple sclerosis (MS) but they are more frequent in neuromyelitis optica (NMO). The objective is to report their presence in a series of cases of NMO and NMO spectrum disorders (NMOSD), as well as to determine their frequency and clinical features.
Patients and MethodsWe conducted a retrospective assessment of medical histories of NMO/NMOSD patients treated in 2 hospitals in Buenos Aires (Hospital Durand and Hospital Álvarez) between 2009 and 2013.
ResultsOut of 15 patients with NMOSD (7 with definite NMO and 8 with limited NMO), 4 presented PPTS (26.66%). PPTS frequency in the definite NMO group was 57.14% (4/7). Of the 9 patients with longitudinally extensive transverse myelitis (LETM), 44.44% (9/15) presented PPTS. Mean age was 35 years (range, 22-38 years) and all patients were women. Mean time between NMO diagnosis and PPTS onset was 7 months (range, 1-29 months) and mean time from last relapse of LETM was 30 days (range 23-40 days). LETM (75% cervicothoracic and 25% thoracic) was observed by magnetic resonance imaging (MRI) in all patients. Control over spasms and pain was achieved in all patients with carbamazepine (associated with gabapentin in one case). No favourable responses to pregabalin, gabapentin, or phenytoin were reported.
ConclusionsPPTS are frequent in NMO. Mean time of PPTS onset is approximately one month after an LETM relapse, with extensive cervicothoracic lesions appearing on the MRI scan. They show an excellent response to carbamazepine but little or no response to pregabalin and gabapentin. Prospective studies with larger numbers of patients are necessary in order to confirm these results.
Los espasmos tónicos paroxísticos dolorosos (ETPD) se definen como espasmos musculares localizados (en uno o más miembros y/o tronco), recurrentes, estereotipados acompañados por dolor intenso y posturas distónicas con una duración aproximada de 20-45 segundos1–7. Estos episodios pueden o no estar desencadenados por movimientos bruscos o estímulos sensitivos y pueden ser clasificados según la localización de la lesión en cerebrales o medulares1–7. Es importante tener en cuenta que los ETPD afectan la calidad de vida, la rehabilitación y las actividades de la vida diaria1 convirtiéndose en un signo que rápidamente debemos pensar, diagnosticar y tratar.
Si bien los ETPD fueron inicialmente descriptos en la esclerosis múltiple (EM) por Matthews en 19581, recientemente se ha publicado una mayor frecuencia en la neuromielitis óptica (NMO)7,8, variando su incidencia de 3-35%1–8 en pacientes con mielopatía desmielinizante de diversas causas (EM, NMO, idiopática).
Con el objetivo de determinar la frecuencia de asociación entre ETPD y NMO y su espectro (NMOSD), las características clínicas y respuesta al tratamiento, analizamos una serie de casos de pacientes que cumplen los criterios para NMO/NMOSD.
Materiales y métodosSe revisaron las historias clínicas, forma retrospectiva, de pacientes que concurrieron a los servicios de neurología en 2 centros de la Ciudad de Buenos Aires (Hospital General de Agudos Dr. Carlos G. Durand y Hospital General de Agudos Dr. Teodoro Álvarez), durante el periodo de enero de 2009 a diciembre 2013.
Este estudio consideró pacientes con NMO definida y NMOSD con anticuerpos antiaquaporina 4 (APQ4) positivo. Se tomó como diagnóstico definido de NMO a los criterios de Wingerchuck et al., 20069, donde se necesita los 2 criterios mayores: mielitis transversa aguda (MTA) más neuritis óptica (NO) y al menos 2 de los siguientes criterios: resonancia magnética (RM) de encéfalo inicial normal o que no cumpla con los criterios de Barkhoff/Tintoré para EM, RM espinal con afección de al menos 3 segmentos medulares (LETM) e IgG-NMO o AQP4 positiva en suero6. Definimos NMOSD según Wingerchuck et al., 200710, a las formas limitadas como la NO aislada bilateral o recurrente, LETM aislada o recurrente, EM óptico espinal, NO o LETM asociada a enfermedades autoinmunes y NO o LETM asociada a lesiones típicas de NMO. Asimismo, definimos ETPD como fue descripta en la introducción del trabajo.
ResultadosSe observaron 15 pacientes con NMO/NMOSD: 7 con NMO definida y 8 con NMOSD limitada: NO recurrente (n:3), NO aislada (n:3) y LETM aislada (n:2), de los cuales 4 presentaron ETDP (4/15: 26,66%). En los pacientes con NMO definida la frecuencia fue del 57,14% (4/7) y ninguno (0/8) en el grupo NMOSD. Tomando en cuenta todos los pacientes con LETM el 44,44% (4/9) presentaron ETPD.
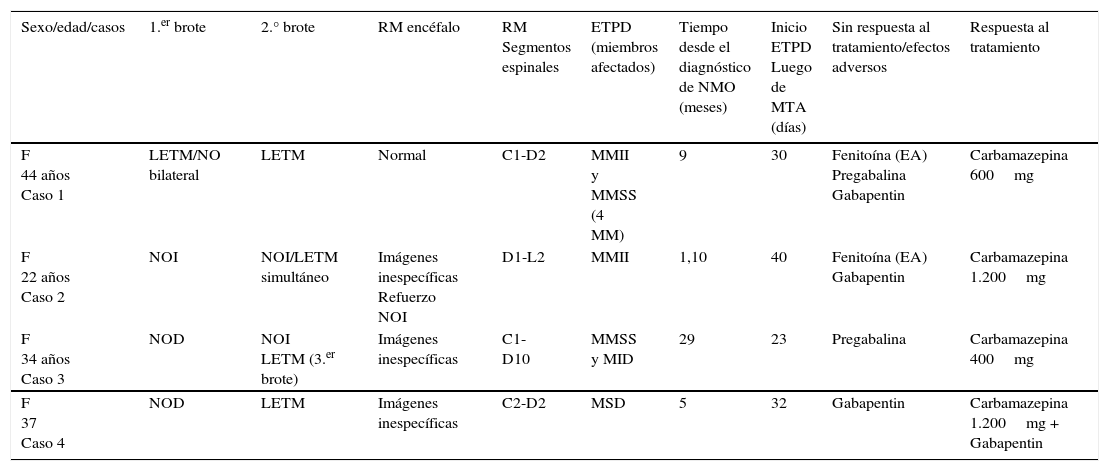
Cabe aclarar que ningún paciente presentó injuria cervicodorsal traumática, traumatismo de cráneo, antecedentes familiares ni historia de distonía, ninguno recibió tampoco medicación antidopaminérgica (por ejemplo: neurolépticos). A continuación se describen brevemente los 4 casos reportados (ver tabla 1).
Características clínicas, radiológicas y de tratamiento de los 4 pacientes con ETPD
| Sexo/edad/casos | 1.er brote | 2.° brote | RM encéfalo | RM Segmentos espinales | ETPD (miembros afectados) | Tiempo desde el diagnóstico de NMO (meses) | Inicio ETPD Luego de MTA (días) | Sin respuesta al tratamiento/efectos adversos | Respuesta al tratamiento |
|---|---|---|---|---|---|---|---|---|---|
| F 44 años Caso 1 | LETM/NO bilateral | LETM | Normal | C1-D2 | MMII y MMSS (4 MM) | 9 | 30 | Fenitoína (EA) Pregabalina Gabapentin | Carbamazepina 600mg |
| F 22 años Caso 2 | NOI | NOI/LETM simultáneo | Imágenes inespecíficas Refuerzo NOI | D1-L2 | MMII | 1,10 | 40 | Fenitoína (EA) Gabapentin | Carbamazepina 1.200mg |
| F 34 años Caso 3 | NOD | NOI LETM (3.er brote) | Imágenes inespecíficas | C1-D10 | MMSS y MID | 29 | 23 | Pregabalina | Carbamazepina 400mg |
| F 37 Caso 4 | NOD | LETM | Imágenes inespecíficas | C2-D2 | MSD | 5 | 32 | Gabapentin | Carbamazepina 1.200mg + Gabapentin |
F: femenino; LETM: mielitis transversa longitudinal extensa; MID: miembro inferior derecho; MM: miembros; MMII: miembros inferiores; MMSS: miembros superiores; MSD: miembro superior derecho; NOI: neuritis óptica izquierda; NOD: neuritis óptica derecha.
Mujer, 44 años, presentó LETM y NO bilateral, recibió tratamiento con metilprednisolona endovenosa (EV) y corticoides vía oral (VO) con mejoría parcial. Presentó recaída a los 9 meses con cuadriparesia severa y lesiones extensas a nivel cervical. Recibió plasmaféresis con mejoría, corticoides y azatioprina. Al mes presenta ETPD de los 4 miembros. Inició fenitoína con mejoría rápida, pero por hepatotoxicidad se suspendió. Escasa respuesta a pregabalina y gabapentin. Mejoró con carbamazepina 600mg/día, se suspende por aumento de las transaminasas. Presenta lenta mejoría en meses.
Caso 2Mujer, 22 años, NO izquierda hace 4 años, presentó LETM dorsal. Mejoró con metilprednisolona EV, corticoides VO y azatioprina. Presentó a los 40 días ETPD progresivos en miembros inferiores (MMII). No respondió a la pregabalina. Se inició fenitoína con excelente respuesta, se suspende al mes por farmacodermia. No responde a gabapentin. Franca mejoría con carbamazepina 1.200mg/día.
Caso 3Mujer, 34 años, NO derecha hace 29 meses, se agregó NO izquierda 8 meses después. Presentó LETM cervicodorsal con trastornos sensitivos en miembros superiores (MMSS) y paraparesia en MMII. Mejoró con metilprednisolona EV, corticoides VO y azatioprina. Presentó a los 23 días ETPD en ambos MMSS y en MI derecho. No respondió a la pregabalina. Franca mejoría a carbazepina 400mg/día.
Caso 4Mujer, 37 años, NO derecha hace 5 meses, se agregó LETM cervicodorsal. Mejoró con metilprednisolona EV, corticoides VO y azatioprina. Presentó a los 30 días ETPD en MSD. Realizó tratamiento con gabapentin con escasa mejoría. Se le agregó carbamazepina 1.200mg/día con franca mejoría.
En resumen, de estos pacientes (n:4) la edad media fue de 35 años (rango: 22-38 años). Cien por cien de sexo femenino. El tiempo transcurrido entre el diagnóstico de NMO y la aparición de los espasmos fue de 7 meses (rango: 1-29 meses) y desde la última recaída de LETM una media de 30 días (rango: 23-40 días). El 100% presentó LETM en RM (cervicodorsal 75% y dorsal 25%). Los ETPD se constataron luego del diagnóstico: media 7 meses (rango: 1-29 meses) y con respecto a la última recaída de LETM la media fue: 30 días (rango: 23-40 días). El 100% presentó LETM en RM (cervicodorsal 75% y dorsal 25%). El 75% presentó ETPD luego del primer ataque de LETM y el 25% (caso 1) luego del segundo ataque. Con respecto al tratamiento el 75% recibió gabapentin y el 50% pregabalina sin respuesta. El 50% recibió fenitoína con buena respuesta pero presentaron efectos adversos que obligaron a su suspensión (farmacodermia y hepatotoxicidad). El 100% presentó control de los espasmos y el dolor con carbamazepina (uno asociado a gabapentin). El 100% recibió tratamiento agudo con metilprednisolona EV. Los 4 pacientes fueron tratados con azatioprina más meprednisona VO como preventivo de las recaídas.
DiscusiónEn este estudio encontramos que de 15 pacientes con NMO/NMOSD 4 presentaron ETPD (26,66%) y el porcentaje aumenta si consideramos solo a los pacientes con LETM (44,44%), y es mayor aún en los pacientes con NMO definida (57,14%). En contraste, datos recientes de la literatura muestran que en la EM menos del 5% presentan ETPD7. Los estudios occidentales sobre EM reportan datos similares (2-10%)4,11, sin embargo, un estudio oriental (Japón) muestra una incidencia mayor (17,18%) en la EM6. Pero estos datos pueden confundir ya que, un alto porcentaje de estos pacientes presentaban desmielinización extensa medular y podrían corresponder a EM variante óptico espinal, entidad que algunos autores consideran dentro del NMOSD. Estos datos sugieren que los ETPD se asocian con mayor frecuencia a la NMO en comparación con la EM. Además, en un estudio se observó que el dolor en la NMO es más severo e incapacitante que en la EM12. Esto puede ser explicado porque a diferencia de la EM, la NMO frecuentemente causa mielitis transversa con severa desmielinización y necrosis medular (tisular) debido a que los autoanticuerpos aquaporina-4 (AQP4) son del tipo IgG1 y provocan citotoxicidad dependiente del complemento. Asimismo, las citoquinas (IL 17, IL 8 y factor estimulante de colonias de granulocitos, entre otras) reclutan neutrófilos y eosinófilos en los espacios perivasculares para su degranulación, causando la muerte astrocitaria4. Esta pérdida conduce a la muerte de los oligodendrocitos, lo que provoca degeneración axonal y muerte neuronal12,13.
Es interesante destacar que ninguna paciente presentó ETPD como inicio de la enfermedad. Una paciente presentó NO asociado a LETM en el inicio (caso 1) y el resto solo NO de inicio. Los ETPD se asociaron a una recaída de LETM con una media de 30 días posteriores al evento. En un estudio se observó que los ETPD se asociaron a la recuperación de un primer episodio de mielitis, lo que sugirió que la remielinización parcial medular tendría un papel importante en el desarrollo de ETPD comparado con la propia desmielinización causada por la NMO7. En otro estudio reciente se observó que la presencia de ETPD luego de un primer ataque de MTA tenía una especificidad del 100% y sensibilidad de 67% para NMO9,14. Estos resultados marcan que los ETPD podrían ser potenciales candidatos para los criterios de soporte luego de una nueva revisión de los criterios actuales9.
La fisiopatología de los ETPD en NMO aún no está del todo clara. Sin embargo, en el estudio realizado por Ostermann et al.11 sobre EM y ETPD se propuso una teoría donde los mediadores químicos inflamatorios (ácido araquidónico, leucotrienos y prostaglandinas) producirían irritación axonal causando este signo. A su vez otro estudio propuso que la afección temprana de las fibras centrípetas en la médula espinal con fibras corticoespinales relativamente indemnes se activaría de forma efáctica con la consiguiente difusión axonal dentro de la lesión parcialmente desmielinizada en los tractos de fibras4,7,11. Ante la frecuencia de que los ETPD sean bilaterales se ha postulado que son el resultado de lesiones a nivel del bulbo raquídeo (decusación del haz piramidal), la médula espinal o ambos7. En nuestra muestra, 3 pacientes (casos 1, 3, 4) presentaron lesiones cervicales altas, 2 de las cuales (casos 1 y 3) presentaron ETPD bilaterales. Sin embargo, también lo presentó una paciente con lesiones dorsales (caso 2). Estudios adicionales que investiguen un número mayor de casos serán necesarios para avalar dicha teoría.
Con respecto al tratamiento nuestras 4 pacientes respondieron de manera excelente a la carbamazepina. Dos pacientes tuvieron buena respuesta a la fenitoína pero presentaron efectos adversos que resultaron en la suspensión de la medicación. De acuerdo a nuestra muestra no se encontraron efectos beneficiosos con gabapentin y pregabalina. Estos resultados coinciden con la mayoría de los reportes aunque serían necesarios estudios controlados que avalen estas conclusiones.
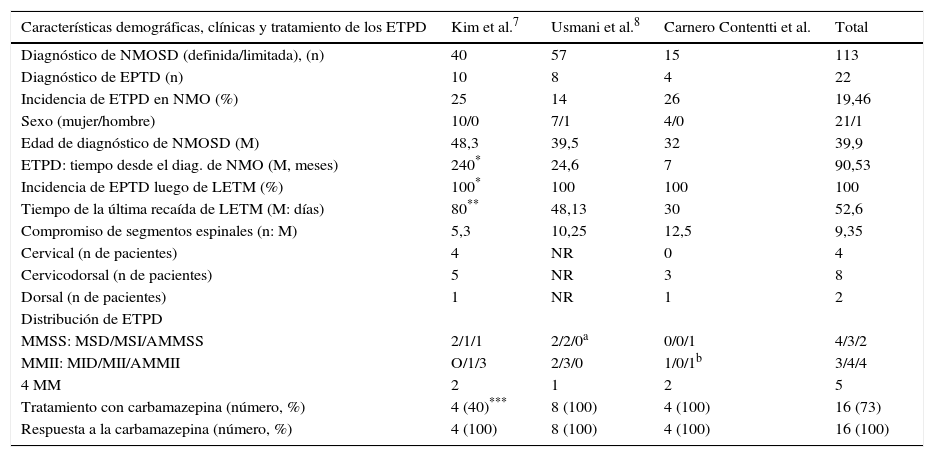
Como limitaciones del estudio podemos nombrar principalmente el número bajo de pacientes y su metodología retrospectiva. Esto determina que cada resultado que obtuvimos deberá ser confirmado con estudios prospectivos con un mayor número de pacientes. Es interesante comentar que hay pocos trabajos publicados (serie de casos)3,7,8,14–16 que hablan específicamente de la asociación de NMO con ETPD con datos similares (ver tabla 2), lo que nos motivó a presentar este trabajo aún con este número pequeño de pacientes.
Comparación de características clínicas, demográficas y tratamiento en 3 series específicas de pacientes con ETPD asociado a NMO
| Características demográficas, clínicas y tratamiento de los ETPD | Kim et al.7 | Usmani et al.8 | Carnero Contentti et al. | Total |
|---|---|---|---|---|
| Diagnóstico de NMOSD (definida/limitada), (n) | 40 | 57 | 15 | 113 |
| Diagnóstico de EPTD (n) | 10 | 8 | 4 | 22 |
| Incidencia de ETPD en NMO (%) | 25 | 14 | 26 | 19,46 |
| Sexo (mujer/hombre) | 10/0 | 7/1 | 4/0 | 21/1 |
| Edad de diagnóstico de NMOSD (M) | 48,3 | 39,5 | 32 | 39,9 |
| ETPD: tiempo desde el diag. de NMO (M, meses) | 240* | 24,6 | 7 | 90,53 |
| Incidencia de EPTD luego de LETM (%) | 100* | 100 | 100 | 100 |
| Tiempo de la última recaída de LETM (M: días) | 80** | 48,13 | 30 | 52,6 |
| Compromiso de segmentos espinales (n: M) | 5,3 | 10,25 | 12,5 | 9,35 |
| Cervical (n de pacientes) | 4 | NR | 0 | 4 |
| Cervicodorsal (n de pacientes) | 5 | NR | 3 | 8 |
| Dorsal (n de pacientes) | 1 | NR | 1 | 2 |
| Distribución de ETPD | ||||
| MMSS: MSD/MSI/AMMSS | 2/1/1 | 2/2/0a | 0/0/1 | 4/3/2 |
| MMII: MID/MII/AMMII | O/1/3 | 2/3/0 | 1/0/1b | 3/4/4 |
| 4 MM | 2 | 1 | 2 | 5 |
| Tratamiento con carbamazepina (número, %) | 4 (40)*** | 8 (100) | 4 (100) | 16 (73) |
| Respuesta a la carbamazepina (número, %) | 4 (100) | 8 (100) | 4 (100) | 16 (100) |
AMMII: ambos miembros inferiores; AMMSS: ambos miembros superiores; diag.: diagnóstico; ETPD: espasmos tónicos paroxísticos dolorosos; LETM: mielitis transversa longitudinal extensa; M: media; MID: miembro inferior derecho; MII: miembro inferior izquierdo; MM: miembros; MMII: miembros inferiores; MMSS: miembros superiores; MSD: miembro superior derecho; MSI: miembro superior izquierdo; n: número; NMOSD: espectro de la neuromielitis óptica; NR: no remitido.
Un paciente presentó en forma simultánea ETPD y diagnóstico de NMO, otro (caso 2) luego 4.490 días y el caso 4 1.963 días posterior al diagnóstico de NMO. Es por ello que la media es de 240 meses, de lo contrario sería de 25,5 días (80%).
El 90% de los episodios de los ETPD ocurrió dentro de los primeros 80 días luego del primer evento de mielitis. La media de ETPD ocurrió 48,13 días luego del primer episodio de LETM (8 de 10 pacientes, 80%).
El 40% realizó tratamiento con fenitoína, el 20% con gabapentin y el 10% con baclofeno con buena respuesta.
Los ETPD se presentan frecuentemente en los pacientes con NMO (57,14% en nuestra serie). Si bien fue descripto desde hace muchos años en la EM, su frecuencia sería menor que en la NMO (<10%). No hemos encontrado la misma asociación en nuestros pacientes con NMOSD limitada dado que estos pacientes presentaron fundamentalmente NO (aislada o recurrente). Los ETPD generalmente aparecen al mes de una recaída de mielitis con lesiones cervicodorsales extensas en RM. No deben considerarse como una recaída de la enfermedad sino como un síntoma paroxístico que debemos reconocer y tratar por su impacto en la calidad de vida. Los ETPD tienen poca o nula respuesta a pregabalina y gabapentin pero excelente respuesta a carbamazepina que consideramos la droga de elección.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

