





El síndrome X frágil es una forma de retraso mental heredado. Es consecuencia de una expansión anormal del número de repeticiones del trinucleótido CGG. Algunos abuelos de estos niños llegan a ser olvidadizos, tienen frecuentes caídas y sufren otros problemas neurológicos. Los investigadores han encontrado una conexión entre el síndrome X frágil y los síntomas neurológicos de los ancianos. Esto ha llevado a reconocer un síndrome inicialmente denominado “temblor intencional, parkinsonismo y atrofia cerebral generalizada en portadores de premutación X frágil”. Al mismo tiempo, en las mujeres, la premutación se ha asociado a fallo ovárico prematuro.
MetodologíaEste artículo revisa la bibliografía acerca de las manifestaciones neurológicas de la premutación X frágil.
ConclusionesLa premutación X frágil supone un riesgo de sufrir trastornos del movimiento y disfunciones cognitivas y debe considerarse en pacientes con una historia familiar de retraso mental o autismo y, particularmente, en mujeres con fallo ovárico prematuro.
Fragile X syndrome is an inherited form of mental retardation. It results from an abnormally expanded number of trinucleotide CGG repeats. Some grandfathers of these children become forgetful, have frequent falls and other neurological problems. Researchers have found a connection between fragile X syndrome and the neurological symptoms in elderly men. This resulted in the recognition of a syndrome originally referred to as “intention tremor, parkinsonism and generalised brain atrophy in carriers of a fragile X premutation”. This premutation is also associated with premature ovarian failure.
MethodologyThis paper reviews the literature on the neurological signs of fragile X premutation.
ConclusionsFragile X premutation is a risk for movement disorders and cognitive dysfunction and it should be considered in patients with a family history of mental retardation or autism, and particularly in those females with premature ovarian failure.
El síndrome del cromosoma X frágil (SXF) es la causa más frecuente de retraso mental heredado y se asocia a un fenotipo físico y conductual bastante característico1. Por su vinculación al cromosoma X, la prevalencia en los varones (1:3.600) es mayor que en las mujeres (1:8.000)2.
Al ser un síndrome de la infancia, se le ha prestado poca atención en la neurología del adulto. Sin embargo, en las últimas décadas se han descrito manifestaciones neurológicas de inicio tardío en familiares de niños con SXF, es decir, en portadores de la enfermedad hasta ese momento asintomáticos.
El SXF se produce por una expansión inestable de repeticiones del triplete citosina-guanina-guanina (CGG) en el gen FMR1(Fragile X Mental Retardation 1 gene) del cromosoma X (Xq27.3)1. En la llamada mutación completa (MCX), causante del SXF, se identifican más de 200 repeticiones de CGG que están anormalmente metiladas bloqueando la producción de la proteína del gen FMR1 (FMRP)1. En la población sana se pueden encontrar entre 5 y 44 repeticiones polimórficas de CGG. Expansiones entre 55 y 200 repeticiones de CGG poseen versiones del gen FMR1 no metiladas que permiten tener valores de FMRP normales o cercanos a la normalidad3. Este intervalo de repeticiones de CGG se denomina premutación X frágil (PXF) y está en mujeres y varones sin manifestaciones del SXF, pero que pueden transmitir el trastorno. Existe una “zona gris”, entre 45 y 54 repeticiones de CGG, que se comporta de forma inestable en la transmisión, pero que podría expandirse a una MCX en dos generaciones4. La transición desde la PXF a la MCX ocurre exclusivamente por transmisión materna, posiblemente durante la oogénesis. El riesgo de transición parece depender enteramente del tamaño de la expansión5,6, y en la actualidad se cuestiona el papel de las interrupciones AGG en la secuencia CGG7. También se han descrito deleciones genómicas como causa menos frecuente de SXF8.
La prevalencia de PXF entre los varones es más baja (1:813)9 que entre las mujeres (1:100)10.
Patogenia y neuropatologíaLa FMRP está implicada en la unión, la estabilidad, el transporte y la transcripción del ARN con repercusiones faciales, esqueléticas, cardiovasculares, endocrinas y, especialmente, del sistema nervioso11, donde interviene en la regulación y transmisión de la información en la sinapsis por medio de mecanismos de inhibición12. El déficit o la disminución de FMRP ocasiona una sobreexpresión de proteínas del citoesqueleto, de especial relevancia en la estructura y plasticidad de las sinapsis y del receptor 5 del glutamato que interviene en la regulación de la excitabilidad de las redes neuronales13.
Los estudios histológicos en la MCX identifican espinas dendríticas de morfología inmadura14 y la formación de agregados de polirribosomas neuronales15 en relación con una disminución del tamaño del vermis posterior cerebeloso16, el núcleo caudado14 y el hipocampo17.
En un primer momento se pensaba que la PXF tenía nulo o escaso efecto en la expresión de FMR1 y, por lo tanto, no se asociaba a manifestaciones fenotípicas. Posteriormente, en sujetos con PXF, se ha demostrado una disminución en la producción de FMRP18 y una elevación de las concentraciones de FMR1 en ARNm, que señalan una escasa eficiencia del gen FMR119 y tienen un efecto citotóxico20 al inducir la acumulación intranuclear de material anómalo en neuronas y células gliales21. Los cuerpos de inclusión se encuentran distribuidos por la corteza y el tronco del encéfalo, con mayor densidad en el hipocampo y en la corteza frontal22, pero también se identifican en las células ganglionares suprarrenales y mesentéricas y en los ganglios de las raíces posteriores y simpáticos paraespinales23. Las inclusiones son positivas para ubiquitina, con negatividad para poliglutamina, tau y sinucleína, y están relacionadas con la degradación de proteínas aún no bien conocidas3. En la PXF, se ha observado una correlación entre el número de inclusiones y el de repeticiones de CGG que podría llegar a ser un potente predictor de afectación neuropatológica24–26. Por el contrario, los pacientes con MCX no tienen cuerpos de inclusión intranucleares27. Más recientemente, se han identificado agregados perinucleares de cadena B de alfa-cristalina que podrían estar relacionados con una predisposición a enfermedades neuroinmunológicas28.
También se ha constatado una pérdida de células de Purkinje, gliosis y afectación de la sustancia blanca, cerebral y cerebelosa, con un patrón diferente del vascular, y espongiosis, particularmente, en los pedúnculos cerebelosos medios29 (tabla 1).
Hallazgos neuropatológicos en la premutación X frágil
| Afección significativa de la sustancia blanca encefálica, con un patrón diferente del vascular, y espongiosis predominantemente en los pedúnculos cerebelosos medios |
| Afección de los astrocitos con abultamiento por las inclusiones |
| Inclusiones intranucleares en el encéfalo y la médula |
En definitiva, el mismo gen tiene dos expresiones opuestas: inactividad en la MCX, que causa un déficit de FMRP y produce un trastorno del neurodesarrollo, o hiperactividad en la PXF, que induce un exceso de ARNm con consecuencias citotóxicas, y condiciona un trastorno neurodegenerativo30,31.
Manifestaciones clínicasLas primeras descripciones de síntomas neurológicos en portadores de PXF se efectuaron en abuelos de niños diagnosticados de SXF y señalaban deterioro cognitivo, temblor y alteraciones en la marcha29. Progresivamente, se han ido recogiendo más manifestaciones neurológicas tardías asociadas a la PXF que ha llevado a definir una entidad clínica propia, actualmente denominada síndrome de temblor/ataxia asociado al X frágil (fragile X-associated tremor/ataxia syndrome [FXTAS])29, que guarda una cierta correlación con los valores de FMRP y los años transcurridos32.
Con todo, las manifestaciones clínicas de la PXF no parecen limitarse al FXTAS; también se ha señalado una mayor incidencia de agresividad, abuso de alcohol y drogas, ansiedad, cuadros obsesivo-compulsivos33 y problemas de aprendizaje34 y se han empezado a describir algunas de las características físicas menores propias del SXF: rasgos faciales, orejas prominentes, hiperflexibilidad de las articulaciones, etc.35. En esta misma línea, hay evidencias de un fenotipo conductual y neuropsicológico asociado a la PXF en adultos, sin manifestaciones clínicas de FXTAS, que aún no está bien definido36.
Inicialmente, en niños con PXF no se observaron déficit en el neurodesarrollo37. Posteriormente, se ha identificado una cierta relación entre valores de FMPR y retraso mental y/o autismo, lo que indica un espectro de gravedad clínica relacionado con el déficit de la proteína35,38, con riesgo de afección cognitiva y conductual39. De hecho, comparados con controles sanos, los niños con PXF tienen una incidencia más alta de retraso en el neurodesarrollo, déficit de atención, problemas de conducta, ansiedad y trastornos del espectro autista40,41.
Durante tiempo, se consideró que las mujeres con PXF no tenían manifestaciones clínicas. Posteriormente, se señaló que cerca del 20% sufría fallo ovárico prematuro42, un fenotipo distintivo con ligeros rasgos físicos propios del SXF35 y una mayor incidencia de síntomas de ansiedad y depresión43. El fallo ovárico prematuro no ocurre en mujeres con MCX, lo que indica que está relacionado con la toxicidad de las concentraciones elevadas de FMR1 en el ARNm presentes, casi exclusivamente, en la PXF3. Con todo, los estudios en mujeres con PXF tienen el problema de la heterogeneidad de las muestras por la gran variabilidad de ratios de activación del gen FMR1.
Síndrome temblor/ataxia asociado al X frágilInicialmente, el FXTAS se definió como un cuadro de temblor intencional progresivo, ataxia cerebelosa y deterioro cognitivo en varones con PXF, de inicio tardío (50-60 años de edad)29 y una prevalencia que aumenta con la edad44.
Posteriormente, se han documentado otras manifestaciones clínicas que incluyen parkinsonismo, neuropatía periférica, disfunción vegetativa, debilidad proximal en las extremidades inferiores, trastornos de conducta y demencia45–48. Al mismo tiempo, se han ido describiendo casos de FXTAS en mujeres, si bien en menor número y con un fenotipo menos severo22, explicable por la presencia de un segundo alelo normal y una inactivación aleatoria del X49.
En 2006, Hall et al50 sistematizaron las principales manifestaciones clínicas del FXTAS que han sido corroboradas por otros autores51,52. La mayoría de los casos tienen trastornos de la marcha (95%), temblor (80%), parkinsonismo (57%) y neuropatía en las extremidades inferiores (60%).
Las dificultades en la marcha son, fundamentalmente, por ataxia cerebelosa acentuadas por el parkinsonismo o/y la neuropatía periférica. La afección cerebelosa se traduce también en alteraciones posturales, dismetría y disartria.
El temblor suele ser intencional (3-5Hz), pero es de reposo en el 10% y mixto en el 30%. Generalmente, se inicia en la mano dominante y, en pocos años, es bilateral. La severidad del temblor contrasta con la levedad de otros síntomas.
El parkinsonismo se expresa como moderada bradicinesia, ligera rigidez en las extremidades superiores y temblor de reposo que, en algunos casos, es indistinguible del parkinsonismo idiopático53.
La neuropatía se manifiesta por debilidad proximal de las piernas, hipoestesia en calcetín alto (en ocasiones, con calambres o quemazón), abolición de los reflejos osteotendinosos y moderada reducción de la velocidad de conducción. En la PXF, con y sin FXTAS, se ha encontrado una correlación inversa entre la velocidad de conducción y los valores de FMR154.
La disfunción vegetativa suele manifestarse como pérdida del control de los esfínteres (53%) e impotencia (80%). La demencia se da en el 20% de los casos, generalmente, asociada a agitación, agresividad, desinhibición y depresión, describiendo un patrón frontosubcortical47.
Habitualmente, la primera manifestación es el temblor y plantea diagnósticos diferenciales55–57. Progresivamente, suele ir apareciendo ataxia de la marcha, parkinsonismo, neuropatía periférica y deterioro cognitivo. En las etapas más tardías, el trastorno puede tener las características clínicas y de neuroimagen de una atrofia multisistémica de tipo cerebelosa58.
En un estudio longitudinal de 12 años, en pacientes con FXTAS, el número de repeticiones de CGG no se relacionó con la progresión del deterioro motor ni cognitivo59. Sin embargo, se ha observado una correlación directa entre el tamaño de las repeticiones de CGG y la intensidad de los déficit motores en los varones, mientras que en las mujeres se limita al grado de ataxia60. Con todo, un estudio reciente61 relaciona pequeñas expansiones de la repetición de CGG con el parkinsonismo. Por otra parte, sólo el 40% de los sujetos con FXTAS tienen antecedentes familiares de enfermedad de Parkinson, demencia o trastornos degenerativos no especificados, lo que indica formas de mutación esporádica o de una penetrancia incompleta62.
En el FXTAS, la resonancia magnética (RM) cerebral ha identificado cambios en la sustancia blanca, prefrontal y cerebelosa, y disminución del volumen del córtex temporal inferior, amígdala-hipocampo y cerebelo63. Habitualmente, se observa un aumento de la señal T2 en los pedúnculos cerebelosos medios y en la sustancia blanca adyacente que tiene valor como criterio de diagnóstico44. En mujeres con FXTAS, se han identificado alteraciones más discretas que en los varones, pero con el mismo patrón64. Por otra parte, hay correlación entre el tamaño de la repetición CGG y el volumen cerebeloso, el aumento del tamaño ventricular y la hiperintensidad de la sustancia blanca en la RM25,65.
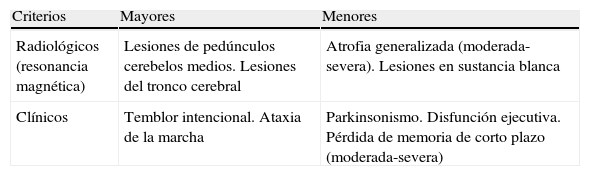
En 2004, el grupo de Jacquemont44 propuso unos criterios diagnósticos para el FXTAS que permitirían diferenciarlo de otros trastornos del movimiento (tablas 2 y 3), que se consolidaron como guía diagnóstica mediante posteriores estudios multidisciplinarios66,67. Con estos criterios, su prevalencia en varones de más de 50 años sería de 1/3.00044. En diferentes muestras de pacientes con temblor esencial, ataxia cerebelosa progresiva esporádica, atrofia multisistémica y parkinsonismo atípico, sin historia familiar de SXF, no se identificó ningún caso de PXF55,68–70. Por lo tanto, en varones adultos con aparición tardía de ataxia, temblor de acción, parkinsonismo, deterioro cognitivo o neuropatía, sólo está justificado el estudio genético de FMR1 si hay historia familiar de retraso mental o trastornos del espectro autista53,71 o cuando la RM es indicativa72. También está justificado en mujeres con esos antecedentes y algunas de las manifestaciones clínicas señaladas de inicio tardío, especialmente cuando han sufrido fallo ovárico prematuro64.
Criterios de diagnóstico del síndrome de temblor/ataxia asociado al X frágil
| Criterios | Mayores | Menores |
| Radiológicos (resonancia magnética) | Lesiones de pedúnculos cerebelos medios. Lesiones del tronco cerebral | Atrofia generalizada (moderada-severa). Lesiones en sustancia blanca |
| Clínicos | Temblor intencional. Ataxia de la marcha | Parkinsonismo. Disfunción ejecutiva. Pérdida de memoria de corto plazo (moderada-severa) |
Probabilidad diagnóstica del síndrome de temblor/ataxia asociado al X frágil
| Diagnóstico | Criterios: inclusión de 55-200 repeticiones de CGG |
| Seguro | 1 signo radiológico mayor+1 síntoma clínico mayor o inclusiones astrocíticas |
| Probable | 1 signo radiológico mayor+1 síntoma clínico menor o 2 síntomas clínicos mayores |
| Posible | 1 signo radiológico menor+1 síntoma clínico mayor |
Algunos autores consideran indicado el estudio de FMR1 en el diagnóstico diferencial de las ataxias espinocerebelosas de varones adultos69,73–75, particularmente, en cuadros oligosintomáticos que muestran una acentuación de la clínica76. También se ha advertido sobre esta posibilidad en mujeres diagnosticadas de esclerosis múltiple con fallo ovárico prematuro y evolución clínica tórpida o atípica77. En nuestro medio, Rodríguez-Revenga et al78 han señalado la necesidad de tener presente esta entidad en el diagnóstico diferencial de la enfermedad de Huntington.
En definitiva, en los pacientes con cuadros clínicos evolutivos de temblor y/o ataxia y/o parkinsonismo atípico y/o afección cognitiva o comportamental se debe interrogar siempre por la historia familiar, buscando no sólo cuadros similares, sino también casos de retraso mental, trastornos del espectro autista y/o menopausia precoz.
ConclusionesLa amplia investigación llevada a cabo en la última década pone de manifiesto la naturaleza dinámica del genotipo y fenotipo X frágil a lo largo de la trayectoria vital, si bien son necesarios más estudios clínicos y neuropatológicos para poder definir mejor sus repercusiones nosológicas.
La PXF se asocia a un amplio espectro de manifestaciones clínicas, especialmente neurológicas en el denominado FXTAS, pero también incluye algunos déficit neuropsicológicos y síntomas neuropsiquiátricos específicos.
Con los conocimientos actuales sobre la PXF es preciso tener presente esta entidad ante determinadas manifestaciones neurológicas en el adulto y hay que procurar indagar siempre sobre antecedentes de menopausia precoz y/o SXF. Al mismo tiempo, es importante lograr una correcta detección del SXF en la infancia que, a su vez, facilitará la identificación temprana de PXF en otros miembros de la familia.
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.

