






La neurofibromatosis tipo 1 es el trastorno neurocutáneo más frecuente. La mayoría de las series de casos publicadas son sobre la población pediátrica.
Material y métodosEstudio transversal de los casos de neurofibromatosis tipo 1 en las consultas de neurología recogidos en una base de datos. Se han analizado las diferentes variables clínicas que conforman el diagnóstico, así como las variables demográficas y neurorradiológicas.
ResultadosSe han encontrado un total de 31 pacientes con neurofibromatosis tipo 1. La edad media ha sido de 28,9 años y el 58,4% son mujeres. Los sujetos con lesiones tipo Unidentified bright objects (UBO) son más jóvenes que los que no las presentan (22,45±8,22 años vs. 32,5±10,64; p=0,011), por el contrario, los sujetos con neurofibromas son mayores que los que no los tienen (30,56±10,68 años vs. 18,25±4,34; p=0,032). No hay diferencias de sexo en la presentación de las variables clínicas ni radiológicas. Siete pacientes presentaron tumores (22,6%), 3 fueron gliomas del tracto óptico (uno de ellos bilateral), 3 neurofibromas plexiformes y un astrocitoma pilocítico del troncoencéfalo.
ConclusionesLos pacientes con neurofibromatosis tipo 1 no solo presentan lesiones tumorales a nivel periférico en forma de neurofibromas, sino también a nivel del sistema nervioso central. La edad de los sujetos que tienen neurofibromas es mayor que la que no los presentan, sin embargo, los que presentan UBO son más jóvenes que los que no poseen estas lesiones.
Type 1 neurofibromatosis is the most common neurocutaneous syndrome. Most published case series study the paediatric population.
Material and methodsCross-sectional study of cases of type 1 neurofibromatosis from neurology departments that were recorded in a database. We analysed the different clinical variables providing the diagnosis as well as demographic and neuroradiological variables.
ResultsWe found a total of 31 patients with type 1 neurofibromatosis. The mean age was 28.9 years and 58.4% were women. Subjects with unidentified bright objects (UBOs) were younger than those without them (22.45±8.22 years vs. 32.5±10.64; P=.011). In contrast, subjects with neurofibromas were older than those without them (30.56±10.68 years vs. 18.25±4.34; P=.032). No sex differences were found in the presentation of clinical or radiological variables. Seven patients (22.6%) had tumours; 3 were optic pathway gliomas (1 bilateral), 3 were plexiform neurofibromas, and 1 was a pilocytic astrocytoma in the brainstem.
ConclusionsPatients with type 1 neurofibromatosis presented both peripheral neurofibromas and tumorous lesions of the central nervous system. Subjects with neurofibromas were older than those who did not present them, while subjects with UBOs were younger than those without such lesions.
La neurofibromatosis tipo 1 (NF1) fue descrita por primera vez en 1882 por von Recklinghausen. Constituye el síndrome neurocutáneo más frecuente, con una incidencia aproximada de uno por cada 3.000 a 3.500 nacidos vivos1. Afecta por igual a ambos sexos. Es una enfermedad hereditaria de transmisión autosómica dominante con alta penetrancia y una gran variabilidad en su expresión clínica. En un 30-50% de los casos corresponden a mutaciones de novo. En 1990 se identificó el gen responsable de la enfermedad en el brazo largo del cromosoma 17, así como la proteína producida por el mismo, la neurofibromina2.
El diagnóstico de la NF1 se basa en la presencia de por lo menos 2 de los criterios establecidos por la National Institute of Health Consensus Development Conference en 1988 (tabla 1), aunque actualmente el diagnóstico se establece por la identificación de la mutación en el gen de la NF1, que se encuentra en el 95% de los casos. Muchas de las manifestaciones de la NF1 tienen una importante relación con la edad, así los neurofibromas no suelen aparecer antes de la adolescencia y su frecuencia aumenta con la edad. Sin embargo, las «manchas café con leche», generalmente están presentes a los 5 años y su número declina a partir de los 50 años3. Hay estudios que revelan que los pacientes con NF1 tienen disminuida su expectativa de vida4.
Criterios diagnósticos de la neurofibromatosis tipo 1. Se requieren la presencia de 2 o más de los siguientes criterios
| 1. Seis o más «manchas café con leche» del siguiente diámetro: |
| ≥5mm antes de la pubertad |
| ≥15mm después de la pubertad |
| 2. Dos o más neurofibromas de cualquier tipo o un neurofibroma plexiforme |
| 3. Pecas axilares o inguinales (signo de Cowden) |
| 4. Glioma del tracto óptico |
| 5. Dos o más nódulos de Lisch (hamartomas benignos del iris) |
| 6. Lesiones óseas típicas: |
| Displasia de esfenoides |
| Displasia o adelgazamiento del cortex del hueso largo (pseudoartrosis) |
| 7. Familiar de primer grado con NF1 |
En pacientes con NF1 se han descrito alteraciones de la señal en imágenes de resonancia magnética nuclear (RMN) potenciadas en T2 conocidas como Unidentified bright objects (UBO) en un 43-93% de los niños5. Los UBO son infrecuentes en pacientes con NF1 por encima de los 20 años. Su localización habitual incluye el globo pálido, tálamo, hipocampo y troncoencéfalo. Aunque la mayoría de las lesiones podrían permanecer estables o incluso desaparecer, algunas pueden transformarse en gliomas6.
Ya que la mayoría de las series de pacientes con NF1 corresponden a poblaciones pediátricas, nuestro objetivo es conocer las características clínicas y neurorradiológicas de una serie de pacientes adultos con NF1.
Material y métodosEstudio trasversal de los pacientes atendidos en consultas de neurología general con diagnóstico de NF1 recogidos en una base de datos y obteniendo los datos de la valoración clínica habitual. El diagnóstico de NF1 se estableció según los criterios de la National Institute of Health Consensus Conference. Los pacientes que son valorados en nuestras consultas son a partir de los 14 años.
Se recogieron los siguientes datos demográficos y clínicos: sexo, edad, antecedentes familiares, procedencia de la consulta, presencia de «manchas café con leche», neurofibromas, efélides axilares o inguinales, nódulos de Lisch, alteraciones esqueléticas y clínica neurológica. Así mismo, se revisaron las pruebas de neuroimagen de los pacientes para establecer la presencia de lesiones tipo UBO5, gliomas de la vía óptica, astrocitomas cerebrales, estenosis del acueducto de Sílvio y lesiones de naturaleza vascular. En los pacientes con lesiones se ha comprobado la evolución de las mismas en las pruebas de neuroimagen seriadas.
Las variables cuantitativas se han expresado en términos de media y desviación estándar, mientras que las cualitativas se han expresado en términos de porcentaje. Se ha estudiado si existen diferencias respecto a la edad o sexo de las diferentes características clínicas y neurorradiológicas mediante la comparación de medias y tablas de contingencias con la prueba de Chi-cuadrado. Se ha utilizado el paquete estadístico SPSS® v. 15.0. La significación estadística se ha establecido para una p<0,05.
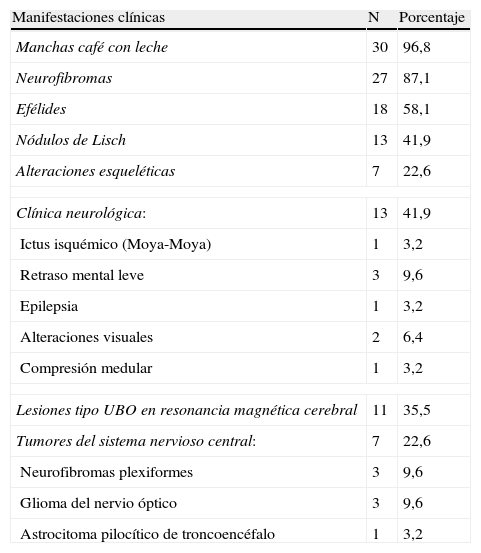
ResultadosSe analizaron un total de 31 pacientes con NF1. La edad media fue de 28,97 años (±10,8), 17 fueron mujeres (54,8%). Se realizó estudio genético en solo 4 pacientes (12,9%). En la tabla 2 se muestran las características clínicas y neurorradiológicas de los pacientes con NF1. Se registraron antecedentes familiares en 19 sujetos (61,3%). La gran mayoría presentaban «manchas café con leche» (96,8%). Se observaron neurofibromas en el 87,1% de los sujetos, efélides en un 58,1% y nódulos de Lisch en un 41,9%. Siete sujetos presentaron alteraciones esqueléticas (22,6%), de ellos 5 tenían escoliosis, una displasia de esfenoides y una macrocrania.
Manifestaciones clínicas de los pacientes con neurofibromatosis tipo 1 (n=31)
| Manifestaciones clínicas | N | Porcentaje |
| Manchas café con leche | 30 | 96,8 |
| Neurofibromas | 27 | 87,1 |
| Efélides | 18 | 58,1 |
| Nódulos de Lisch | 13 | 41,9 |
| Alteraciones esqueléticas | 7 | 22,6 |
| Clínica neurológica: | 13 | 41,9 |
| Ictus isquémico (Moya-Moya) | 1 | 3,2 |
| Retraso mental leve | 3 | 9,6 |
| Epilepsia | 1 | 3,2 |
| Alteraciones visuales | 2 | 6,4 |
| Compresión medular | 1 | 3,2 |
| Lesiones tipo UBO en resonancia magnética cerebral | 11 | 35,5 |
| Tumores del sistema nervioso central: | 7 | 22,6 |
| Neurofibromas plexiformes | 3 | 9,6 |
| Glioma del nervio óptico | 3 | 9,6 |
| Astrocitoma pilocítico de troncoencéfalo | 1 | 3,2 |
Trece pacientes (41,9%) presentaron manifestaciones clínicas neurológicas, de ellos uno tuvo un ictus isquémico a los 43 años secundario a síndrome de Moya-Moya, 5 tuvieron cefalea tipo migraña de aparición en la edad juvenil, 3 presentaron retraso mental leve, uno tuvo epilepsia con crisis parciales complejas con generalización secundaria, 2 tuvieron alteraciones visuales con déficit de la agudeza visual por la lesión tumoral del nervio óptico y un paciente presentó un cuadro medular tipo Brown-Sequard por compresión por neurofibroma a nivel dorsal. Todos los pacientes fueron estudiados con RMN cerebral. En 19 pacientes se repitió la RMN cerebral al menos en una ocasión. Se observaron lesiones tipo UBO en 11 sujetos (35,5%). En 2 de los 3 pacientes con retraso mental se evidenciaron lesiones tipo UBO. Se constató una disminución de las lesiones tipo UBO en 4 pacientes seguidos durante más de 5 años con RMN cerebral de repetición.
Finalmente, 7 pacientes presentaron tumores (22,6%), 3 fueron gliomas del tracto óptico (fig. 1) (uno de ellos bilateral), 3 tuvieron neurofibromas plexiformes y uno presentó astrocitoma pilocítico del troncoencéfalo (fig. 2). Excepto un neurinoma plexiforme y el astrocitoma pilocítico que aparecieron en las pruebas de neuroimagen de control a los 7 y 10 años del diagnóstico, el resto de tumores se evidenció en la primera prueba de imagen realizada en la edad pediátrica. En ambos casos fueron un hallazgo buscado al aparecer clínica neurológica.


Resonancia magnética cerebral en secuencia T1 con contraste y plano axial, donde se evidencia una tumoración heterogénea, expansiva a nivel de protuberancia que capta contraste a nivel periférico. Los hallazgos corresponden a un tumor pilocítico de troncoencéfalo en un paciente con neurofibromatosis tipo 1.
No se encontraron diferencias de edad respecto a la presencia o ausencia de «manchas café con leche», efélides, nódulos de Lisch, alteraciones esqueléticas ni tumores. Al analizar la variable lesiones tipo UBO se encontró una diferencia respecto a la edad, de modo que la edad media fue significativamente menor en aquellos sujetos que presentaban estas lesiones (22,45±8,22 años vs. 32,5±10,64; p=0,011). Por otro lado, la variable neurofibromas también mostraba diferencias respecto a la edad, siendo la edad media de los sujetos con neurofibromas significativamente superior respecto a los que no los presentaban (30,56±10,68 años vs. 18,25±4,34; p=0,032).
No se encontraron diferencias en las variables estudiadas respecto al sexo. En cuanto a la procedencia de los sujetos, 21 estaban diagnosticados por pediatría, en el resto: 7 fueron consultados por clínica dermatológica en sujetos con antecedentes familiares de NF1 conocida y 3 fueron un hallazgo exploratorio al remitir por clínica neurológica (2 migrañas y un glioma de nervio óptico).
DiscusiónEn nuestra serie, el hallazgo clínico más frecuente fue la presencia de «manchas café con leche» y únicamente un sujeto tuvo menos de las 6 manchas necesarias para el diagnóstico. Mientras que en algunas series se ha documentado que el número de «manchas café con leche» disminuye a partir de los 50 años, nosotros no hemos llegado a esta conclusión probablemente por el escaso número de pacientes con esa edad.
Los neurofibromas cutáneos generalmente aparecen después de la preadolescencia y afectan a la mayoría de los adultos con NF17. En nuestra serie hemos corroborado cómo los sujetos sin neurofibromas son más jóvenes, lo que implica indirectamente que aparecen con la edad. Son lesiones benignas que no suelen sufrir transformación maligna8. Las efélides que se localizan a nivel de axilas e ingles tienen una frecuencia del 58% similar a otras series9. Los nódulos de Lisch son hamartomas bilaterales benignos del iris y son patognomónicos de la NF1. No está claro si su incidencia aumenta con la edad9. Para su diagnóstico se requiere el examen con lámpara de hendidura por parte de un oftalmólogo familiarizado con la NF1.
Como en otras series, la escoliosis que afecta a una sexta parte de los pacientes10 aparece en la infancia, es idiopática y no suele requerir tratamiento quirúrgico. Otras alteraciones esqueléticas descritas en la NF1 solo se han descrito puntualmente en nuestra serie como la displasia de esfenoides y la macrocrania.
Resaltamos que 3 de nuestros pacientes (10%) presentaron retraso mental leve, apareciendo en la mayoría lesiones tipo UBO. Aunque no se pueden extraer conclusiones por el escaso tamaño muestral, otros autores ya han relacionado la presencia de lesiones tipo UBO con el rendimiento cognitivo11. La presencia de sintomatología neurológica como la migraña y la epilepsia parece una asociación casual7, ya que su prevalencia es similar a la población general (16 y 3% respectivamente). Otros estudios han relacionado la existencia de epilepsia con la presencia de lesiones tumorales intracraneales, pero no con lesiones tipo UBO12. Nuestra única paciente con crisis tenía una lesión isquémica en el contexto de un síndrome de Moya-Moya.
La presencia de lesiones tipo UBO en la resonancia es del 35,5% inferior a lo publicado en otras series pediátricas5,12,13. Estudios transversales han asociado la presencia de lesiones tipo UBO con diferentes patrones de la NF1, como los gliomas ópticos, los nódulos de Lisch y los neurofibromas14. A diferencia de los neurofibromas que parecen aumentar con la edad, las lesiones tipo UBO van desapareciendo5,15. Los datos extraidos de nuestro análisis se encuentran en consonancia, porque en nuestra serie de sujetos adultos la prevalencia fue menor, existiendo diferencias de edad entre los sujetos con lesiones tipo UBO que eran más jóvenes, que aquellos que no las presentaban.
La aparición de tumores cerebrales en nuestra serie es similar a otras previas con gliomas del nervio óptico en torno al 10% y de astrocitoma pilocítico de troncoencéfalo del 3%16,17. En varios de nuestros pacientes se ha evidenciado una disminución de las lesiones tipo UBO en la resonancia magnética, sin embargo, no podemos extraer conclusiones generales dado que el seguimiento ha sido irregular y carecemos de muchas pruebas de neuroimagen antiguas. Sin embargo, en nuestra serie los gliomas ópticos y el neurofibroma plexiforme han permanecido estables a lo largo del tiempo. El paciente con estenosis del acueducto de Silvio presentaba un glioma del nervio óptico con colocación de válvula de derivación a los 5 años, siendo esta asociación frecuentemente descrita en niños con NF118. Aunque los tumores cerebrales en la NF1 suelen ser benignos, debemos sospechar malignidad en aquellos de localizaciones inusuales o que se comporten de manera agresiva19.
Actualmente existe consenso entre los expertos que la NF1 no tiene solamente lesiones tumorales a nivel periférico como los neurofibromas, sino también a nivel del sistema nervioso central. Esta incidencia de tumores sobre la población general sugiere realizar un estudio de neuroimagen con resonancia cerebral en sujetos con NF1 que presentan clínica neurológica, aunque no está claro hasta qué edad deben hacerse estudios seriados. Nuestra sugerencia es realizar estudios de imagen de control en los pacientes con UBO para corroborar su disminución de tamaño con la edad, así como ante la aparición de focalidad neurológica u oftalmológica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

