



El conocimiento del alcance socioeconómico de la patología neuromuscular es esencial para la planificación de recursos y la concienciación social.
DesarrolloSe ha realizado una revisión de los datos publicados hasta el momento sobre epidemiología, mortalidad, dependencia e impacto sociosanitario de la esclerosis lateral amiotrófica y las enfermedades neuromusculares en España. Además, se ha recogido cómo está organizada la atención neurológica en estos pacientes.
ConclusionesLa patología neuromuscular constituye un grupo muy heterogéneo de enfermedades, algunas de las cuales se consideran raras por su baja frecuencia. Esta patología supone entre el 2,8 y el 18% de los motivos de consulta en un Servicio de Neurología. En España, las cifras de prevalencia e incidencia de esclerosis lateral amiotrófica son similares a otros países; sin embargo, se desconoce el número de pacientes con otras enfermedades neuromusculares. Son enfermedades crónicas, progresivas y debilitantes, lo que condiciona una importante discapacidad y dependencia. Esto repercute directamente en los costes sanitarios y sociales asociados a la enfermedad. Se ha calculado que el coste de un paciente con esclerosis lateral amiotrófica o enfermedad de Duchenne se acerca a los 50.000 euros anuales. La patología neuromuscular tiene una gran complejidad etiológica, diagnóstica y pronóstica, y requiere un manejo multidisciplinar. Las Unidades especializadas deben ser las encargadas del seguimiento de estos pacientes.
A thorough knowledge of the socioeconomic scope of neuromuscular disease is essential for managing resources and raising social awareness.
DevelopmentOur group reviewed current data on the epidemiology, mortality and dependence rates, and socioeconomic impact of amyotrophic lateral sclerosis and neuromuscular diseases in Spain. We also recorded how neurological care for these patients is organised.
ConclusionsNeuromuscular disorders are a very heterogeneous group of diseases, and some are very rare. These disorders account for between 2.8% and 18% of the total motives for a neurological consultation. In Spain, prevalence and incidence figures for amyotrophic lateral sclerosis are similar to those in other countries; however, figures for patients with other neuromuscular diseases are not known. Since the diseases are chronic, progressive, and debilitating, they cause considerable disability and dependence, which in turn directly affects healthcare and social costs associated with the disease. The costs generated by one patient with amyotrophic lateral sclerosis or Duchenne disease have been calculated at about 50 000 euros per year. Neuromuscular disease shows aetiological, diagnostic, and prognostic complexity, and it requires multidisciplinary management. Follow-up for these patients should be entrusted to specialised units.
La Fundación del Cerebro trabaja para la divulgación y la concienciación de la sociedad sobre la situación real que viven los pacientes neurológicos y su entorno más cercano. Su fin último es mejorar el bienestar de los afectados y para ello ha establecido diversos objetivos orientados a la información, prevención, investigación, asistencia e integración. Como parte del programa de concienciación, la Fundación realiza informes de impacto social sobre las diferentes patologías neurológicas en España.
En este trabajo se aborda específicamente la patología neuromuscular. Es un grupo de enfermedades muy amplio y complejo, en el que se agrupan entidades que por sus peculiaridades merecerían un análisis independiente. Este informe está dedicado a la esclerosis lateral amiotrófica (ELA) y otras enfermedades neuromusculares (ENM) «no ELA» de especial relevancia, a las que nos referiremos con las siglas ENM para facilitar la exposición.
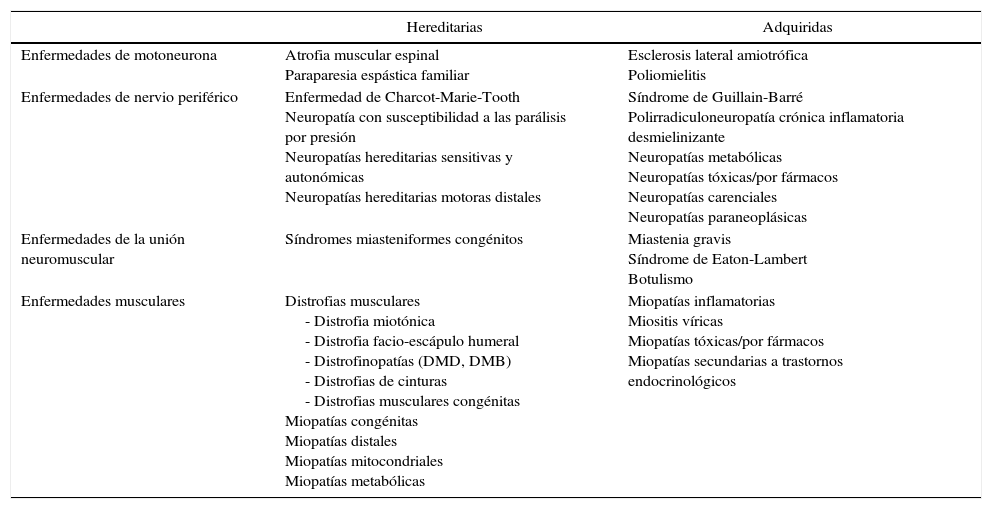
La patología neuromuscular es aquella que afecta al sistema nervioso periférico, al músculo esquelético, a la unión neuromuscular y a la médula espinal. El conjunto de trastornos es muy numeroso y viene marcado por la heterogeneidad (tabla 1). Se clasifican en función de la localización de la lesión: enfermedades de motoneurona, radiculopatías, plexopatías, neuropatías, enfermedades de la unión neuromuscular y, por último, enfermedades musculares. Dentro de cada grupo, el origen de la enfermedad puede ser metabólico, infeccioso, tóxico, inmunomediado, genético o neurodegenerativo. Su comienzo acontece tanto en la infancia como en la edad adulta, y el curso evolutivo es variable: agudo, subagudo o crónico. El diagnóstico y el tratamiento de la patología neuromuscular requieren una alta especialización, por lo que su estudio y manejo constituyen una subespecialidad dentro la neurología general.
Principales enfermedades neuromusculares en función de su localización
| Hereditarias | Adquiridas | |
|---|---|---|
| Enfermedades de motoneurona | Atrofia muscular espinal Paraparesia espástica familiar | Esclerosis lateral amiotrófica Poliomielitis |
| Enfermedades de nervio periférico | Enfermedad de Charcot-Marie-Tooth Neuropatía con susceptibilidad a las parálisis por presión Neuropatías hereditarias sensitivas y autonómicas Neuropatías hereditarias motoras distales | Síndrome de Guillain-Barré Polirradiculoneuropatía crónica inflamatoria desmielinizante Neuropatías metabólicas Neuropatías tóxicas/por fármacos Neuropatías carenciales Neuropatías paraneoplásicas |
| Enfermedades de la unión neuromuscular | Síndromes miasteniformes congénitos | Miastenia gravis Síndrome de Eaton-Lambert Botulismo |
| Enfermedades musculares | Distrofias musculares - Distrofia miotónica - Distrofia facio-escápulo humeral - Distrofinopatías (DMD, DMB) - Distrofias de cinturas - Distrofias musculares congénitas Miopatías congénitas Miopatías distales Miopatías mitocondriales Miopatías metabólicas | Miopatías inflamatorias Miositis víricas Miopatías tóxicas/por fármacos Miopatías secundarias a trastornos endocrinológicos |
DMB: distrofia muscular de Becker; DMD: distrofia muscular de Duchenne.
La ELA, junto a sus variantes (esclerosis lateral primaria, atrofia muscular progresiva y parálisis bulbar progresiva), es la enfermedad de neurona motora más frecuente del adulto. Se trata de una enfermedad debilitante, progresiva, de carácter neurodegenerativo, secundaria a la afectación de las neuronas motoras superiores (localizadas en la corteza motora precentral) e inferiores (localizadas en los núcleos motores del tronco y el asta anterior de la médula espinal). Aunque típicamente aparece de forma esporádica (un caso aislado en una familia), entre un 5 a un 10% de los casos presentan agrupamientos familiares, la mayoría de las veces siguiendo un patrón de herencia autosómica dominante (pero también descritas con un patrón autosómico recesivo y ligado al sexo dominante). Un 5-10% de los casos presenta una demencia asociada, en general del tipo fronto-temporal, que puede preceder, aparecer simultáneamente o posteriormente al inicio de la ELA. Pero hasta en más de un 50% de los casos los pacientes muestran rasgos de disfunción disejecutiva en los estudios neuropsicológicos.
La enfermedad tiene un curso progresivo e inexorable que lleva a la muerte, típicamente, en menos de 5 años desde el inicio, en general, como consecuencia de una insuficiencia respiratoria restrictiva progresiva. No existe un tratamiento curativo y el tratamiento desde su inicio está basado en medidas paliativas de control de síntomas.
El conjunto de las ENM distintas de la ELA es muy diverso, pero estas entidades comparten una serie de peculiaridades que generan problemas similares y justifican su consideración unitaria. La manifestación clínica más común es la pérdida de fuerza, que con frecuencia es progresiva. La debilidad conlleva problemas ortopédicos secundarios, como rigidez o deformidades articulares, que son especialmente importantes en las enfermedades que se inician en la edad pediátrica. Otros síntomas frecuentes en la esfera motora son la fatiga, las contracturas o la dificultad para la relajación muscular. Las principales manifestaciones no motoras son la alteración de la sensibilidad, el dolor y la disautonomía. Con la evolución de la enfermedad, es frecuente la aparición de problemas respiratorios y/o cardíacos, que constituyen la principal causa de mortalidad, y deglutorios, que condicionan la necesidad de gastrostomía. Son, por tanto, trastornos que requieren un manejo multidisciplinario en el que intervienen diferentes especialistas de forma coordinada.
Existen ENM frecuentes, como las polineuropatías adquiridas, que afectan al 8% de la población general. Pero muchas de ellas son enfermedades raras. Estas vienen definidas por una prevalencia que no supera el 5 por 10.000 en la Unión Europea (Reglamento 141/2000 de la Comisión Europea). Aunque la frecuencia aislada de cada una resulte muy baja, la suma de todas ellas alcanza una cifra considerable y se calcula que en España puede haber un total de 3 millones de enfermos raros. Las ENM raras están en su mayoría determinadas genéticamente y pueden comenzar en la infancia. Existen tratamientos sintomáticos para las complicaciones derivadas de la enfermedad, pero la mayoría no tiene un tratamiento curativo, por lo que habitualmente siguen un curso crónico, progresivo y gravemente discapacitante. La baja prevalencia de estas enfermedades condiciona un desconocimiento de las mismas no solo en la sociedad, sino también en la comunidad médica, lo que provoca una indeseable demora diagnóstica. En España, en los últimos años, se ha potenciado la investigación epidemiológica y fisiopatológica de estas enfermedades a través de la actividad del Grupo de Estudio de Enfermedades Neuromusculares de las Sociedad Española de Neurología (SEN), del desarrollo del portal Orphanet y de la creación del Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). Este último cuenta con programas de investigación en medicina genética en el que se incluyen ENM del tipo distrofias musculares, atrofia muscular espinal y enfermedad de Charcot-Marie-Tooth (CMT). De todo lo anterior resulta fácil deducir el enorme impacto individual y social de esta patología, por lo que este informe se centrará especialmente en aspectos relacionados con ENM raras.
EpidemiologíaEpidemiología de la esclerosis lateral amiotróficaLa ELA es la tercera enfermedad neurodegenerativa en incidencia, tras la demencia y la enfermedad de Parkinson. Existen múltiples publicaciones sobre las características epidemiológicas de la ELA que incluyen series poblacionales, estudios de unidades especializadas o de áreas geográficas de alta incidencia. En general, los estudios en la población occidental muestra una incidencia que oscila entre 1 a 2 casos por 100.000 habitantes y año, salvo en las regiones de alta incidencia (isla de Guam, península de Kii, población de Irian Jaya, tribu de Anguru en Australia o la población de Guadalupe en el Caribe). También existen datos sobre la incidencia y prevalencia en distintas razas. Así, estudios multirraciales muestran una incidencia mayor en la población blanca que en la mestiza1, o una incidencia menor en la población afroamericana2. Se estima que la ELA afectará a una persona de cada 400-800, si vive lo suficiente.
La prevalencia, medida dependiente de la supervivencia y, por lo tanto, sujeta a modificaciones con el desarrollo de nuevas formas de tratamiento, es baja por la alta mortalidad de la enfermedad, oscilando en los distintos estudios entre 2 a 5 casos por 100.000 habitantes (aunque existen cifras mayores y menores).
Existen pocos estudios epidemiológicos en la población española. Estos incluyen la población de Cantabria3, Segovia4, la isla de la Palma5 y, más recientemente y de mayor calidad, en Cataluña6. En los primeros, la incidencia es algo inferior, pero en el estudio más reciente la incidencia y prevalencia es similar a otros estudios (1,4 y 5,4 por 100.000, respectivamente).
Las causas genéticas también presentan una distribución universal, aunque existen mutaciones concretas más frecuentes en algunas áreas, por un efecto fundacional (expansión C9orf72 en Cerdeña o Finlandia, mutación p.D90A del gen SOD1 recesiva en Suecia, mutación p.A4V del gen SOD1 en Estados Unidos). La edad media de inicio se encuentra entre los 60-69 años, con un pico de incidencia a los 70-75 años y una disminución de la incidencia en edades superiores (a diferencia de lo que ocurre con la enfermedad de Parkinson o con la demencia tipo Alzheimer). En todas las series occidentales, la incidencia en los varones es ligeramente superior a la de las mujeres, pero en los estudios en afroamericanos esta diferencia se invierte. En cuanto a la forma de inicio por localización, un tercio de los pacientes se inician de forma bulbar, con otros 2/3 de forma espinal (repartidos equitativamente entre inicio en los miembros superiores o inferiores). Un pequeño grupo de casos presenta un inicio generalizado o respiratorio.
Con estos datos, la estimación de pacientes con ELA en España es de 3 casos nuevos de ELA al día, presentando en este momento ELA en España más de 3.000 personas. Dado que el pico de incidencia es algo menor que en las otras enfermedades neurodegenerativas, esto se traduce en que más del 50% de los casos afecta a personas en edad laboral, plenamente productivas.
Epidemiología de las enfermedades neuromuscularesLa multitud de ENM dificulta su abordaje global y existen pocos estudios epidemiológicos en los que se incluyan todas o la mayoría de ellas. En este sentido, el trabajo publicado por Emery en 1991 continúa siendo una referencia7. En él revisó más de 150 publicaciones sobre la frecuencia de varias ENM hereditarias que afectan a adultos y a niños. Consideró las distrofias musculares (distrofia muscular de Duchenne [DMD], distrofia muscular de Becker [DMB], distrofias de cinturas y distrofia facio-escápulo-humeral [DFSH]), las enfermedades miotónicas (enfermedad de Steinert y miotonías congénitas), la atrofia muscular espinal y las neuropatías sensitivo-motoras hereditarias. El autor estimó una prevalencia global para ambos sexos de 286 casos por millón de habitantes, por lo que uno de cada 3.500 individuos podría desarrollar una ENM incapacitante a lo largo de su vida. Durante la década de los 90, se publicaron algunos estudios epidemiológicos europeos de base poblacional. En Örebro, Suecia, la prevalencia puntual de las ENM, incluyendo enfermedades de motoneurona, neuropatías hereditarias, enfermedades con miotonía, distrofias musculares y miositis, se calculó en 84 por 100.000 habitantes8. En Irlanda del Norte se restringió el análisis a las ENM hereditarias, cuya prevalencia se estimó en 34,5 por 100.000 habitantes (1 de cada 2.900)9. Si sólo se considera la edad pediátrica, la estimación de la prevalencia en menores de 20 años en Bolonia fue de 24,9 por 100.000 habitantes10, mientras que en la región oeste de Suecia la cifra ascendía a 63,1 por 100.000 habitantes para todas la ENM y 53,1 por 100.000 si solo se consideraban las enfermedades hereditarias11.
Respecto a la situación en España, se desconoce la prevalencia del conjunto de las ENM. La Federación ASEM, que agrupa a 21 asociaciones de pacientes afectados por ENM, estima que hay más de 60.000 enfermos en nuestro país.
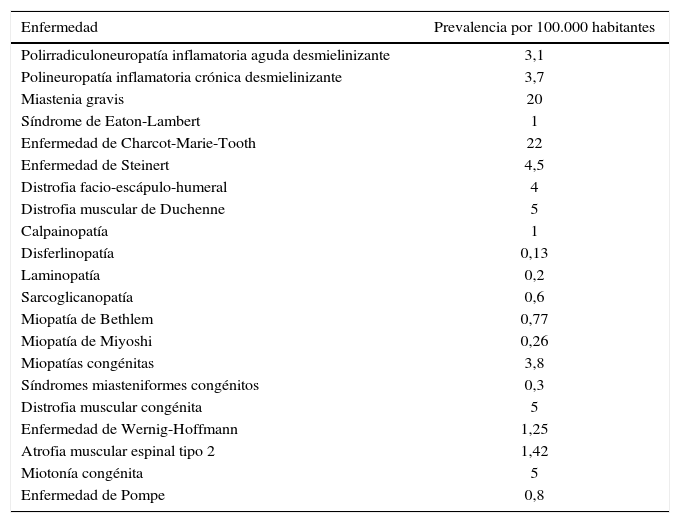
Si en vez de considerar el conjunto de las ENM, se analiza enfermedad por enfermedad, sí es posible obtener más resultados. Orphanet es el portal de información de referencia en enfermedades raras y medicamentos huérfanos en Europa. En mayo del 2014 publicó una estimación de la prevalencia de las enfermedades raras en Europa tras realizar una búsqueda sistemática en Medline, páginas web, registros de pacientes y libros médicos12. Las cifras referentes a las ENM se recogen en la tabla 2. Es una información valiosa, pero los autores del estudio reconocen que tiene varias limitaciones. Hay un bajo grado de consistencia entre las fuentes consultadas y con frecuencia existe confusión entre la prevalencia y la incidencia. Además, puede haber una sobreestimación de cifras puesto que muchos trabajos se realizan en áreas geográficas de alta prevalencia para una determinada patología o se basan en datos hospitalarios. Sorprende también que en el listado de enfermedades no figure la DMB o la enfermedad de Mc Ardle.
Estimación de la prevalencia de las ENM (Orphanet Report Series, mayo del 2014)
| Enfermedad | Prevalencia por 100.000 habitantes |
|---|---|
| Polirradiculoneuropatía inflamatoria aguda desmielinizante | 3,1 |
| Polineuropatía inflamatoria crónica desmielinizante | 3,7 |
| Miastenia gravis | 20 |
| Síndrome de Eaton-Lambert | 1 |
| Enfermedad de Charcot-Marie-Tooth | 22 |
| Enfermedad de Steinert | 4,5 |
| Distrofia facio-escápulo-humeral | 4 |
| Distrofia muscular de Duchenne | 5 |
| Calpainopatía | 1 |
| Disferlinopatía | 0,13 |
| Laminopatía | 0,2 |
| Sarcoglicanopatía | 0,6 |
| Miopatía de Bethlem | 0,77 |
| Miopatía de Miyoshi | 0,26 |
| Miopatías congénitas | 3,8 |
| Síndromes miasteniformes congénitos | 0,3 |
| Distrofia muscular congénita | 5 |
| Enfermedad de Wernig-Hoffmann | 1,25 |
| Atrofia muscular espinal tipo 2 | 1,42 |
| Miotonía congénita | 5 |
| Enfermedad de Pompe | 0,8 |
En este informe de impacto social no es factible revisar cada una de las ENM, pero sí se analizarán en mayor profundidad algunas de las más representativas: las 3 distrofias musculares más frecuentes (enfermedad de Steinert, DFSH y DMD), el CMT, el síndrome de Guillain-Barré (SGB) y la miastenia gravis (MG).
Distrofia miotónica. Enfermedad de SteinertLa distrofia miotónica (DM) es la distrofia muscular más frecuente en la edad adulta. Se transmite con herencia autosómica dominante. Es una enfermedad multisistémica caracterizada por afectación muscular, en forma de debilidad y miotonía, cataratas, afectación cardíaca, problemas gastrointestinales y trastornos hormonales13. A nivel mundial, la prevalencia oscila entre 2,1 a 14,3 por 100.000 habitantes, según las diferentes poblacionales. Las cifras de prevalencia más elevadas se han documentado en Saguenay-Lac-Saint-Jean, en la región de Quebec, con 158 casos por 100.000 habitantes en 201014. En España se publicaron, a principios de los años 90, 2 artículos sobre la epidemiología de la DM en nuestro país. Burcet et al. estimaron la prevalencia de la DM en la isla de Mallorca en 11 casos por 100.000 habitantes. Utilizaron como fuente los historiales médicos hospitalarios15. Poco después, López de Munáin et al. estudiaron la provincia de Guipúzcoa, encontrando una prevalencia que llegaba a los 26,5 casos por 100.000 habitantes. Es una de las cifras más elevadas a nivel mundial y los autores la justifican tanto por razones metodológicas (exhaustividad en la búsqueda de casos y consulta de varias fuentes) como por un posible efecto fundador local, similar a la situación de Saguenay-Lac-Saint-Jean16.
Distrofia facio-escápulo-humeralEs una distrofia muscular que se caracteriza por debilidad de predominio en los músculos faciales y de la cintura escapular, y afectación asimétrica. Se transmite con herencia autosómica dominante. Tras la DM, es una de las distrofias musculares más frecuentes. Emery recogía en 1991 una prevalencia en países occidentales que oscilaba entre 2,2 y 66,9 casos por millón de habitantes7. Por citar datos más recientes de un país mediterráneo como el nuestro, en Italia un estudio poblacional realizado en la provincia de Padua estimó la prevalencia en 44 casos por millón de habitantes17.
En España no hay datos de prevalencia de la enfermedad. En la Encuesta sobre Discapacidad, Autonomía personal y situaciones de Dependencia de 2008 no se recoge como tal la DFSH, pero sí figuran como grupo las «distrofias musculares», con 138.200 pacientes entre los 6 y los 64 años18.
DistrofinopatíasLas distrofinopatías ocupan un lugar relevante dentro de las ENM. Son enfermedades con herencia recesiva ligada al cromosoma X. La alteración genética que provoca la ausencia de distrofina genera la DMD, mientras que si queda distrofina residual la enfermedad resultante es la DMB. Tradicionalmente, la DMD se ha designado como la enfermedad genética muscular más frecuente de la infancia, con una incidencia estimada de 1:3.500 varones7. La incidencia de la DMB es menor, 5.4:100.000, y su prevalencia de 2,8:100.000. Frente a estas cifras clásicas, Mah et al.19 han publicado en 2014 un metaanálisis sobre la epidemiología de las distrofinopatías. Los autores revisaron 31 trabajos y dentro de los estudios de base poblacional solo se incluyeron los realizados en Norteamérica y en Europa. La prevalencia estimada para la DMD y DMB fue de 4,78 y 1,53 por 100.000 varones, respectivamente. La incidencia no se pudo analizar por la escasez y la disparidad de los estudios, pero en la DMD oscilaba entre 10,71 (en Italia) y 27,78 (en Canadá) por 100.000 recién nacidos varones al año.
En España no hay datos de prevalencia ni de incidencia. Se dispone de los datos de «distrofias musculares» ya referidos de la Encuesta sobre Discapacidad, Autonomía personal y situaciones de Dependencia de 200818.
Enfermedad de Charcot-Marie-ToothEsta enfermedad engloba un grupo genéticamente heterogéneo de neuropatías hereditarias con afectación motora y sensitiva. Es una de las ENM hereditarias más comunes, con una prevalencia que puede llegar a 40 casos por 100.000 habitantes20. En España se llevó a cabo un estudio pionero, en el que de forma prospectiva (años 1974-1984) se analizaba la frecuencia de CMT en Cantabria. El diagnóstico de la enfermedad se realizaba entonces sobre la base de las manifestaciones clínicas, el patrón de herencia y los hallazgos neurofisiológicos. La prevalencia en 1985 fue de 28,2 casos por 100.000 habitantes21. Posteriormente, se han publicado series españolas descriptivas que reúnen un número muy importante de casos. Aunque no son de base poblacional, estos estudios son relevantes porque ponen de manifiesto las características clínicas y genéticas propias de los pacientes con CMT de una determinada región22,23.
Síndrome de Guillain-BarréEs una polirradiculoneuropatía aguda inflamatoria de causa inmunitaria. Frente a la gran mayoría de polineuropatías, se trata de una enfermedad de rápida instauración. En un metaanálisis del 2011 sobre la incidencia del SGB, se revisaron todos los trabajos publicados entre 1966-2009; finalmente se analizaron 16, resultando una incidencia anual de 1,11 casos (rango 0,81-1,89) por 100.000 habitantes24. En España, un estudio de referencia demostró en Cantabria una incidencia anual de un caso por 100.000 habitantes entre 1975-198825. Posteriormente, ha persistido en nuestro país el interés por la vigilancia de esta patología a través del Grupo de Estudio Epidemiológico del SGB, en el que han participado 11 centros. Este grupo analizó de forma retrospectiva el SGB en mayores de 20 años entre los años 1985 y 1997, encontrando una incidencia de 0,85 casos por 100.000 habitantes y año. Esta cifra mostraba una tendencia a incrementarse con la edad y a lo largo de los años26. Desde 1996, el análisis pasó a ser prospectivo, con un período de seguimiento mínimo de un año. Entre los años 1998 y 1999 la incidencia ascendió a 1,26 casos anuales por 100.000 habitantes, resultados similares a otros países europeos occidentales27.
Miastenia gravisEs una enfermedad autoinmue de la unión neuromuscular que se caracteriza por debilidad y fatigabilidad. Según un metaanálisis publicado en 2010 que recogía 55 estudios desde 1950 hasta 2007, la incidencia de la MG es de 5,3 casos (rango 1,7-21,3) por millón de habitantes al año y la prevalencia de 77,7 (rango 15-179) casos por millón de habitantes28. A lo largo de los años, se evidencia un aumento de ambas variables de frecuencia debido a factores biológicos, a una mejor identificación de los pacientes y a un cambio en la historia natural de la enfermedad gracias a los avances terapéuticos. En España se han realizado 2 estudios epidemiológicos de base poblacional. En la isla de La Palma, Villagra-Cocco y Villagra-Cocco estimaron en 1996 la prevalencia de la MG en 8,58 casos por 100.000 habitantes29. Aragonès et al. realizaron un estudio prospectivo (1991-2000) en el área de Osona, provincia de Barcelona, y encontraron una incidencia anual de 21,27 casos por millón de habitantes. Como dato relevante, la incidencia aumentaba con edad, hasta alcanzar la cifra de 63,38 casos por millón de habitantes y año en mayores de 65 años. Esto apoyaría la hipótesis de un aumento paradójico de la autoinmunidad en el anciano30.
Como ha quedado reflejado, en nuestro país se dispone de muy poca documentación respecto al número total de enfermos neuromusculares y cuál es la frecuencia de cada patología, por lo que resulta prioritario desarrollar un registro nacional de pacientes. Con esta premisa se emprendió en España en 2010 un proyecto de generación de bases de datos de pacientes con patología neuromuscular. En él se integró una masa crítica de investigadores en ENM de 7 comunidades autónomas, pertenecientes a 6 grupos de CIBERNED, un grupo CIBERER y 6 grupos asociados. La finalidad del registro fue crear una red que eliminara la fragmentación existente y permitiera realizar un esbozo de la distribución de las diferentes patologías en el territorio nacional, lo cual es fundamental para la planificación sanitaria de las ENM. La explotación de estas bases de datos permitiría desarrollar iniciativas de investigación tanto clínica como básica, unificar y validar métodos de diagnóstico, diseñar algoritmos diagnóstico-terapéuticos y realizar estudios terapéuticos prospectivos. Este es un registro complejo, pero está basado en un modelo holandés, que con éxito ha logrado establecer un flujo de información entre los profesionales implicados en esta patología y ha permitido el desarrollo de estudios de excelencia. En la actualidad, la Base de Datos de Patología Neuromuscular ha logrado registrar a más de 5.000 pacientes afectados de diferentes enfermedades y ha posibilitado la realización de trabajos multicéntricos y coordinados en las patologías más prevalentes, como la ELA31. Gracias a esta iniciativa, nuestros pacientes han logrado tener una mayor representación en proyectos europeos, tanto a nivel epidemiológico como participando en ensayos clínicos, al formar parte del consorcio internacional TREAT-NMD Neuromuscular Network32.
MortalidadMortalidad de la esclerosis lateral amiotróficaLa ELA es una enfermedad mortal en un breve plazo. Desde el inicio de la enfermedad, la mitad de las personas con ELA fallece en menos de 3 años, un 80% en menos de 5 años y la mayoría (más del 95%) en menos de 10 años. En un estudio de mortalidad en España (con datos de mortalidad entre 1951 y 1990) se muestra una tendencia al incremento de la misma con los años. La mortalidad global calculada en este estudio es de 1,49 por 100.000, siendo algo más alta en varones (1,90 en varones y 1,21 para mujeres), con un pico entre los 60-69 años33.
Mortalidad de las enfermedades neuromuscularesGran parte de las ENM acortan la esperanza de vida y la muerte puede acontecer incluso en la infancia. El fallo ventilatorio y la cardiopatía (estructural o arritmógena) son las principales causas de fallecimiento.
La DMD es el paradigma de ENM de curso inexorable y fatal, con una historia natural bien conocida. Afortunadamente, su curso se ha modificado en los últimos años gracias a la instauración de un tratamiento multidisciplinario precoz, que incluye la administración de corticoides y el adecuado manejo respiratorio, cardíaco, nutricional y ortopédico. En series históricas, los pacientes fallecían poco después de la adolescencia, pero con la introducción de la ventilación no invasiva y la sistematización de la cirugía de escoliosis la supervivencia se ha prolongado hasta la tercera (y, excepcionalmente, hasta la cuarta) década de la vida34.
Frente a las ENM de evolución más «crónica», existen otras susceptibles de descompensarse rápidamente o sufrir reagudizaciones potencialmente graves. Es el caso de la MG y el SGB. En la MG la situación más grave es la crisis miasténica. Consiste en un fallo respiratorio agudo que requiere ventilación asistida. Hace 50 años la mortalidad asociada a la crisis miasténica podía superar el 30%35. Hoy día esta proporción se ha reducido de forma drástica gracias a la introducción de inmunomoduladores para el manejo crónico de la enfermedad y a la asistencia especializada intensiva en las complicaciones agudas. Esto ha resultado en un aumento de las cifras de prevalencia. En el metaanálisis ya mencionado sobre la epidemiología de la MG, se revisaron 7 estudios que aportaban tasas de mortalidad, obteniendo unas cifras de 0,06 a 0,89 por millón de habitantes y año28. En Estados Unidos, Alshekhlee et al. realizaron un estudio retrospectivo para calcular la mortalidad exclusivamente hospitalaria de pacientes con MG (años 2000-2005). Se incluyó a 5.502 pacientes ingresados en diversos centros, de los cuales el 2,2% falleció en el hospital; esta proporción ascendía al 4,47% en el contexto de una crisis miasténica. Los factores predictivos de muerte fueron la edad avanzada y el fallo respiratorio36. Respecto al SGB, la mortalidad se ha estimado habitualmente entre el 1 y el 5% de los pacientes, debido a complicaciones respiratorias y/o cardiovasculares. El mismo grupo de Alshkhlee publicó un trabajo sobre la mortalidad intrahospitalaria asociada al SGB, con una metodología similar al estudio de MG (años 2000-2004). Se revisó a 4.954 pacientes, de los cuales el 2,58% falleció en el hospital, sin producirse cambios significativos a lo largo de los años. El principal factor predictivo de muerte fue la intubación orotraqueal37.
En España es posible acceder a los datos del Instituto Nacional de Estadística para obtener la tasa de mortalidad de las distintas enfermedades y conocer sus causas. Pero para ello es preciso que en el certificado de defunción se especifique el diagnóstico de la enfermedad (en este caso de la ENM) según la codificación CIE-10 y esta circunstancia no siempre se cumple. En el listado de defunciones por causa de muerte del 2012 figuran cifras absolutas de fallecimientos de las siguientes enfermedades (sin cálculo de tasas): «trastornos miotónicos» 134, «distrofias musculares» 101, «miastenia gravis» 91, «síndrome de Guillain-Barré» 45 y «neuropatía hereditaria motora y sensorial» 638.
Discapacidad y dependenciaLa discapacidad es una limitación funcional para las actividades de la vida diaria debida a una deficiencia y su consecuencia es la dependencia. En la patología neuromuscular se consideran deficiencias la debilidad, las contracturas articulares, la escoliosis, la alteración de la función cardiorrespiratoria, el dolor y la afectación cognitiva. Ello provoca discapacidad en forma de disminución de la movilidad, dificultad para la realización de las actividades de la vida diaria, aumento de la fatiga, problemas cognitivos/del aprendizaje y mala adaptación psicosocial. Las consecuencias de esta discapacidad son menores oportunidades educativas y laborales, mayor dependencia y menor calidad de vida39,40. La dependencia implica la necesidad de asistencia o cuidados por parte de un tercero, por lo que la figura del cuidador es trascendental. Si la enfermedad comienza en la infancia o la adolescencia, la carga familiar y social es prácticamente de por vida.
En la Encuesta Nacional sobre Discapacidad y Dependencia de 2008, 2 grupos de ENM figuran entre las principales causas de discapacidad: la DM y la ELA. Son las únicas categorías diagnósticas referentes a patología neuromuscular, mientras que la encuesta sí identifica otros trastornos neurológicos más reconocidos y homogéneos: Parkinson, accidentes cerebrovasculares, demencia tipo Alzheimer y otras demencias, esclerosis múltiple, daño cerebral adquirido, lesión medular, autismo, síndrome de Down y parálisis cerebral18.
Discapacidad y dependencia en la esclerosis lateral amiotróficaLa ELA, una enfermedad debilitante que lleva a una paralización progresiva del paciente hasta su parálisis completa, es sin duda un modelo de enfermedad que conlleva una gran carga de discapacidad y dependencia. A lo largo de la enfermedad, en un corto plazo, los pacientes pasan de una situación de normalidad a una de dependencia completa. La evolución incide tanto en la capacidad funcional de la persona que el principal método para medir la evolución de la enfermedad y controlar la posible eficacia en ensayos terapéuticos en la actualidad es una escala de incapacidad (la ALSFRS-R), en la que se gradúa la progresión del paciente según las limitaciones que presenta en 4 grupos de funciones (bulbares, movimientos finos, movimientos groseros y respiración). Desde el inicio de la enfermedad, los pacientes ya presentan cierto grado de incapacidad, que se incrementa progresivamente a lo largo de la evolución.
Discapacidad y dependencia en las enfermedades neuromuscularesEn las ENM las limitaciones motoras son las más graves y evidentes. En el ámbito médico existen escalas que permiten evaluar la progresión de la enfermedad. Unas son aplicables a las ENM en general, como Motor Function Measure, Neuromuscular Score, la escala de Brooke y Vignos e incluso una selección de ítems de la CIF, mientras que otras son específicas para una determinada patología, como North Star (para DMD) o Hammersmith Motor Ability Store (para atrofia muscular espinal)41,42. Pero existen otras deficiencias no motoras que merman significativamente la funcionalidad, como el dolor o la afectación cognitiva. El dolor crónico constituye una parte importante de la «carga de enfermedad» en las ENM43,44. La presencia de dolor neuropático está bien documentada en las neuropatías, pero gran número de ENM asocia dolor de diversa naturaleza. Guy-Coichard et al. valoraron la presencia de dolor crónico en pacientes con diversas ENM (DMD, DM, DFSH, miopatías metabólicas y MG) a través de un cuestionario con escalas específicas45. Contestaron 511 pacientes, de los cuales 331 (67,3%) habían presentado dolor en los 3 meses previos. La intensidad del dolor variaba según las enfermedades, pero era máximo en las miopatías metabólicas y en la MG.
La afectación cognitiva está presente en determinadas ENM, como la DMD, la DM y las distrofias musculares congénitas que asocian malformaciones cerebrales. La discapacidad que genera es común a otras enfermedades neurológicas con afectación intelectual, pero esta comorbilidad altera aún más la funcionalidad de los enfermos neuromusculares e incrementa su situación de dependencia.
Impacto económicoLa discapacidad y la dependencia asociadas a la patología neuromuscular tienen unas implicaciones en el gasto sociosanitario evidentes. La ELA y la mayoría de las ENM son enfermedades crónicas y debilitantes, con una carga económica muy alta tanto para el Estado como para las familias. Los pacientes están sometidos a revisiones médicas y sesiones de fisioterapia periódicas, así como a eventuales ingresos hospitalarios ante reagudizaciones o procesos intercurrentes. Habitualmente, precisan ayudas técnicas y ortopédicas, que en ocasiones no están incluidas en el catálogo de material ortoprotésico financiado por la Seguridad Social. La asistencia de un cuidador para la realización de las actividades de la vida diaria suele ser la norma y es frecuente la necesidad de adaptar la vivienda o el medio de transporte. Hay que considerar también los cambios en la vida laboral del enfermo y del cuidador, con la reducción de ingresos correspondiente.
Para calcular los costes globales, hay que considerar los costes directos y los indirectos. Los costes directos incluyen la asistencia médica, el cuidado ambulatorio del paciente y los tratamientos, mientras que los indirectos engloban la disminución de la productividad laboral del paciente o de los cuidadores, bien por absentismo o por mortalidad prematura. Además, existen costes intangibles que hacen referencia al grado de sufrimiento del paciente por la disminución de su calidad de vida.
Impacto económico de la esclerosis lateral amiotróficaEn España no existen estudios que midan el impacto económico de la ELA, pero recientemente se han publicado varios trabajos en otros países sobre este tema. Así, en un estudio irlandés, se calculó que el coste mensual de la enfermedad, desde el momento del diagnóstico hasta la muerte, era de 1.795,21 euros, de los que el 21% se debían a costes asociados al funcionamiento de las unidades multidisciplinares, el 72% a los gastos de los cuidados necesarios basados en la comunidad y el 7% a los gastos derivados de la adquisición de distintas ayudas técnicas y aparataje46. Estos costes se incrementaban en los pacientes con una evolución rápida de la enfermedad o en aquellos en los que se realizó gastrostomía o ventilación mecánica no invasiva. A estos gastos hay que añadir los derivados de otros cuidados informales y los derivados de la pérdida de productividad, que no fueron objeto del estudio.
Un trabajo canadiense cuantificó los gastos directos de pacientes con ELA y sus familias en 32.337 dólares anuales, de los que 19.574 eran pagados por los propios pacientes o sus familias. Los gastos más importantes eran debidos a adecuación del domicilio, ayudas para la movilidad, gastos médicos y de cuidadores privados. También calcularon los costes indirectos por pérdida de ingresos en 56.821 dólares anuales47. Otro trabajo similar en Estados Unidos encontró que el coste total por paciente a lo largo de la enfermedad era de 1.433.992 dólares (85% costeados por las aseguradoras, 9% por la familia y 6% por asociaciones). Los gastos más importantes eran los derivados de cuidadores a domicilio (669.150 dólares), ventilación (212.430 dólares) y cuidados hospitalarios (114.558 dólares)48. Resulta muy interesante un trabajo holandés en el que se compararon los costes mensuales de un grupo de pacientes atendidos en unidades multidisciplinares específicas frente a otro atendido por los recursos generales. Los resultados mostraron que tan solo existe una pequeña diferencia entre ambos (1.336 euros en unidades multidisciplinares frente a 1.271 con cuidados con recursos generales)49.
Finalmente, en un trabajo reciente realizado en Estados Unidos, Larkindale et al. calcularon los gastos de la ELA en 63.693 dólares anuales y los compararon con los gastos en pacientes con DMD y DM (50.952 y 32.236 dólares, respectivamente). Según este estudio, los costes anuales en Estados Unidos de la ELA ascienden a 1.023 millones de dólares, frente a los 787 millones de la DMD y a los 448 millones de la DM50.
Hay que considerar la influencia que esta carga financiera (y la posibilidad de afrontarla) tiene en la toma de decisiones en el cuidado de los pacientes.
Impacto económico de las enfermedades neuromuscularesLos estudios del impacto económico de las ENM son escasos y se centran en las enfermedades más discapacitantes, como las distrofias musculares y, concretamente, la DMD. Ouyang et al. estudiaron en Estados Unidos los costes generados en 2004 por pacientes jóvenes diagnosticados de una distrofia muscular, sin especificar el subtipo51. Se estudió solo a menores de 30 años con aseguradora privada y los gastos médicos fueron de 20.467 dólares por paciente y año. Si solo se consideraba la franja de edad entre 15 y 19 años, la cifra ascendía a 34.161 dólares, probablemente porque en ese grupo etario habría una proporción importante de pacientes con DMD evolucionada y mayores requerimientos asistenciales.
En un estudio más reciente, Landfeldt et al. abordaron los gastos de la DMD en 4 países: Alemania, Italia, Reino Unido y Estados Unidos52. La información se obtuvo a partir de un cuestionario que contestaron 770 pacientes/cuidadores. Los gastos directos por enfermedad y paciente en 2012 se estimaron entre 23.920 y 54.270 dólares. Los gastos indirectos por pérdida de productividad laboral, del paciente o del cuidador, oscilaron entre 18.220 y 21.550 dólares. Por último, los costes intangibles, entendidos como calidad de vida, se calcularon en 37.980-46.080 dólares. En resumen, la carga económica total de una paciente con DMD se estimó en 58.440-71.900 dólares. La cifra más baja correspondía a Italia, probablemente por una organización sanitaria diferente de la de los otros 3 países.
Otras ENM también han despertado interés desde el punto de vista del gasto sociosanitario. Schepelmann et al. estudiaron la MG y la distrofia DFSH en Alemania mediante un cuestionario al que respondieron 41 pacientes con MG y 20 con DFSH53. En 2009 los costes totales por paciente se calcularon en 26.240 euros para la DFSH y 14.950 euros para la MG. La mayor parte se debía a los gastos del seguro médico y a la pérdida de la productividad laboral. Los costes aumentaban en relación con la gravedad de la enfermedad y la necesidad de asistencia personal.
En España disponemos de muy poca información sobre el impacto económico de las ENM. En 2012 se publicó un estudio financiado conjuntamente por el IMSERSO y el Ministerio de Sanidad, Política Social e Igualdad sobre los costes y la calidad de vida de pacientes con enfermedades raras54. Entre ellas se encontraba la DMD. La información se recogió a partir de un cuestionario en el que participaron 57 pacientes. La edad media era de 13,14 años (rango 3-15 años) y el 87,7% precisaba de un cuidador. Los costes medios ascendieron a 94.171 euros, distribuidos de la siguiente manera: costes directos sanitarios 13.828 euros, costes directos no sanitarios 79.312 euros y costes indirectos 1.031 euros. Los gastos en su mayor parte eran debidos a cuidados «informales» o no profesionales (familiares y amigos) (79.243 euros), asistencia médica ambulatoria (4.211 euros) y material sociosanitario (3.496 euros).
Atención neurológica de los pacientes con patología neuromuscularDesde el punto de vista asistencial, en nuestro país se calcula que la patología neuromuscular supone entre el 2,8 y el 18% de los motivos de consulta en un Servicio de Neurología55-57. Es una patológica compleja tanto desde el punto de vista diagnóstico, por su diversidad etiológica, como terapéutico, pues requiere un abordaje multidisciplinario. Si se trata de enfermedades hereditarias además se exige una identificación precoz para facilitar un consejo genético adecuado. Todo ello requiere una alta especialización, por lo que el manejo de estas enfermedades constituye una subespecialidad dentro la neurología general. La SEN cuenta con un Grupo de Estudio específico y abundan las revistas y congresos científicos monográficos.
En este contexto, la atención médica que proporciona un neurólogo sin experiencia en esta patología puede llevar al enfermo a experimentar situaciones de desinformación, angustia y falta de confianza en los servicios sanitarios, que no dan respuesta a qué es lo que le pasa. Esto conduce, en ocasiones, a un auténtico peregrinaje médico buscando segundas opiniones, circunstancia no exenta de un número importante de trámites burocráticos y administrativos. Las unidades clínicas dedicadas al manejo de la ELA/ENM están compuestas por un equipo experto de neurólogos que coordina el resto de especialidades que participan en el seguimiento/tratamiento del paciente neuromuscular: neumología, cardiología, rehabilitación, traumatología, nutrición, genética, fisioterapia, servicios sociales, psiquiatría, etc. Habitualmente compaginan la labor asistencial con la docencia y la investigación.
Atención neurológica de los enfermos con esclerosis lateral amiotróficaEn España, el proceso diagnóstico de la ELA es comparable con el de otros países occidentales del entorno, con un retraso hasta el diagnóstico similar. La sanidad pública dispone de medios para la atención de estos pacientes con la puesta en marcha durante los últimos años de Unidades Multidisciplinares para el manejo de pacientes con ELA en la mayoría de las autonomías. En estas unidades también se genera investigación y se realizan ensayos clínicos internacionales.
Atención neurológica de los enfermos con enfermedades neuromuscularesEn nuestro país estos pacientes son atendidos dentro de una consulta de neurología general en la mayoría de los centros, o bien en una consulta especializada en ENM, que funciona como una Unidad; a ella llegan los enfermos derivados desde otras consultas neurológicas o, con menor frecuencia, desde Atención Primaria. Lo deseable es que sean estas consultas especializadas las encargadas del seguimiento de los pacientes, pero la situación real española muestra importantes carencias. Las diferencias geográficas son notables y hay pacientes que deben desplazarse de su comunidad autónoma a otra para poder ser atendidos en una Unidad experta en ENM. Tampoco están desarrolladas las consultas de transición para el paso del paciente del ámbito pediátrico a las consultas de adulto. Para ayudar a los enfermos, a sus familias y a los profesionales a situar dónde se encuentran los centros especializados en el diagnóstico y el tratamiento de esta patología, la ASEM, en colaboración del Real Patronato de Discapacidad, elaboró en 2012 el primer «Mapa de recursos sanitarios para la atención de las enfermedades neuromusculares»58. En él se detalla por comunidades quiénes forman el equipo profesional, cuáles son los servicios asistenciales implicados, qué tipo de población es atendida (pediátrica y/o adulta) y si hay participación en líneas de investigación o redes colaborativas.
Desde el Grupo de Estudio de Enfermedades Neuromusculares de las SEN se recomienda que exista una Unidad especializada por cada millón de habitantes y que todos los hospitales de referencia dispongan de una59. Se espera que los servicios especializados en la atención integral de esta patología, que ya existen en varias comunidades autónomas, lleguen a ser oficialmente reconocidos por el Comité de Designación de Centros, Servicios y Unidades de Referencia del Sistema Nacional de Salud (SNS).
AsociacionismoLas asociaciones desempeñan un papel muy importante en la orientación y apoyo a los pacientes y en el fomento de la investigación con fines terapéuticos. En España, el movimiento asociativo está consolidado (ANEXO. 1).
Asociaciones de esclerosis lateral amiotróficaEn 1990 se funda la primera asociación a nivel nacional de ELA (Asociación de enfermos de ELA, ADELA). Posteriormente se han desarrollado asociaciones de ELA, y/o delegaciones de la asociación nacional, en las distintas comunidades autónomas. Además, se han creado diversas fundaciones para la promoción de la investigación en ELA, como FUNDELA, la Fundación Diógenes, la Fundación Miquel Valls y la Plataforma afectados de ELA.
Asociaciones de enfermedades neuromuscularesLa Federación ASEM cuenta con más de 8.000 socios y lleva más de 3 décadas dando a conocer las ENM. Ha publicado una Guía de las enfermedades neuromusculares. Información y apoyo a los familiares60. Otras asociaciones igualmente relevantes son la Fundación Isabel Gemio, para la investigación en las distrofias musculares y otras enfermedades raras, Duchenne Parent Project España, centrado en DMD y BMD, Asociación Miastenia de España, Fundación Atrofia Muscular Espinal-FUNDAME o FEDER en el campo de las enfermedades raras.
Las nuevas tecnologías han permitido el desarrollo de portales de Internet, que facilitan y agilizan el intercambio de información. Junto a Orphanet, orientado a las enfermedades raras, hay que destacar TREAT-NMD Neuromuscular Network, iniciativa europea que aúna recursos para pacientes y profesionales. En ella se pueden encontrar guías clínicas de manejo para determinadas ENM (algunas de ellas en español), información sobre registros de pacientes o una actualización sobre los proyectos de investigación y ensayos clínicos en marcha.
Conclusiones- –
Se calcula que en España la patología neuromuscular supone entre el 2,8 y el 18% de los motivos de consulta en un Servicio de Neurología.
- –
En la Encuesta Nacional sobre Discapacidad y Dependencia de 2008, 2 grupos dentro de esta patología figuran entre las principales causas de discapacidad en la población general: la ELA y la DM.
- –
La ELA es la enfermedad de neurona motora más frecuente del adulto. Tiene un curso progresivo e inexorable que lleva a la muerte, típicamente, en menos de 5 años desde el inicio. En nuestro país, la incidencia y la prevalencia de la ELA es de 1,4 y 5,4 por 100.000 habitantes, respectivamente. Se estiman 3 casos nuevos de ELA al día y se calcula que en España hay en este momento más de 3.000 enfermos.
- –
Frente a la relativa homogeneidad de la ELA, el resto de las ENM constituyen un grupo muy amplio y heterogéneo, lo que dificulta su abordaje global. Muchas de estas enfermedades son infrecuentes y se engloban en el conjunto de las enfermedades raras, hecho que difumina las particularidades de la patología neuromuscular.
- –
En España se desconoce el número total de pacientes con ENM por falta de datos. La Federación ASEM estima que la cifra podría alcanzar los 60.000 enfermos. La creación en 2010 de un registro activo de pacientes debe contribuir a mejorar esta situación. En la actualidad, la Base de Datos de Patología Neuromuscular ha logrado registrar a más de 5.000 pacientes afectados de diferentes enfermedades y ha posibilitado la realización de trabajos multicéntricos y coordinados en las patologías más prevalentes.
- –
La ELA y la mayoría de las ENM son enfermedades crónicas, progresivas y debilitantes, lo que condiciona una importante discapacidad y dependencia. Esto repercute directamente en los costes sanitarios y sociales asociados a la enfermedad. Cuanto mayor sea la discapacidad/dependencia, los gastos serán mayores y los índices de calidad de vida relacionados con la salud más bajos. Se ha calculado que el coste de un paciente con ELA o con DMD se acerca a los 60.000 dólares anuales.
- –
La patología neuromuscular tiene una gran complejidad etiológica, diagnóstica y pronóstica, y requiere un manejo multidisciplinar. Las Unidades especializadas deben ser las encargadas del seguimiento de estos pacientes. En nuestro país, esta forma de organizar la asistencia es ya una realidad en el caso de la ELA. Desafortunadamente, no ocurre lo mismo para el resto de ENM. Hay diferencias territoriales notables, por lo que es muy necesario que se reconozcan oficialmente centros de referencia dentro del SNS.
- –
La actividad de las asociaciones de pacientes con ELA/ENM está plenamente consolidada en nuestro medio. Desempeñan un papel clave en la orientación, el apoyo y la mediación de los enfermos y sus familias.
Este trabajo no ha recibido financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Asociación ADELA: http://sites.adelaweb.com/web-adela/
Fundación Fundela: http://www.fundela.info/
Fundación Miquel Valls: http://www.elacat.org/es/
Fundación Diógenes: http://www.fundacionela.com/
Plataforma afectados de ELA: http://www.plataformaafectadosela.org/
Asociación Española de Paraparesia Espástica Familiar: http://www.aepef.org/
Federación ASEM: http://www.asem-esp.org/
Fundación Isabel Gemio: http://www.fundacionisabelgemio.com/
Duchenne Parent Project España: https://www.duchenne-spain.org
Asociación Miastenia de España: http://www.miasteniagravis.es/
Fundación Atrofia Muscular Espinal, Fundame: http://www.fundame.net/
FEDER: http://www.enfermedades-raras.org/
Orphanet: http://www.orpha.net/consor/cgi-bin/index.php?lng=ES/
TREAT-NMD Neuromuscular Network: http://www.treat-nmd.eu/

