






La distonía que responde a la dopa (DRD) engloba a un grupo de enfermedades neurológicas que se presentan con distonía de miembros inferiores y una excelente respuesta a levodopa. La asociación de parkinsonismo es habitual y cada vez se describen más casos de mutaciones en el gen GCH1 sin distonía clínicamente indistinguibles de la enfermedad de Parkinson (EP) idiopática, en los que se demuestra degeneración de la vía nigroestriada. Esta variabilidad fenotípica se ha descrito, incluso, dentro de los miembros de una misma familia.
MétodoDescripción clínica y genética de 14 pacientes que presentan mutaciones asociadas a la DRD.
ResultadosDe las 4 familias, el gen GCH1 está involucrado en 3 de ellas y el gen SPR en una. De los 11 individuos en cuyas familias se detectó mutación de GCH1, 2 presentan fenotipo de DRD, 5 de EP y 4 son asintomáticos. El paciente con mutación de SPR presenta clínicamente distonía combinada con parkinsonismo. En 2 de los pacientes con EP se demuestra degeneración de la vía nigroestriada mediante DaTscan®. En 2 familias se observa variabilidad fenotípica de una misma mutación en GCH1.
ConclusionesLas mutaciones asociadas a la DRD tienen una expresión clínica heterogénea. Las mutaciones en el gen GCH1 pueden presentarse con fenotipo EP en la que se demuestra degeneración de la vía nigroestriada. El estudio del gen GCH1 puede ser útil en los casos de EP sugestivos de origen genético o con antecedentes familiares de distonía.
Dopa-responsive dystonia (DRD) includes a group of neurological disorders that manifest with lower limb dystonia and an excellent response to levodopa. Parkinsonism is commonly associated with DRD, and many cases of GCH1 gene mutations without dystonia but with evidence of nigrostriatal pathway degeneration, indistinguishable to idiopathic Parkinson's disease (PD), have been recently described. This phenotypic variability has been observed even among members of the same family.
MethodClinical and genetic description of 14 patients from 4 different families with DRD associated mutations.
ResultsThe results showed GCH1 mutations in three of the four families, while one family had a mutation in the SPR gene. Within patients with GCH1 mutations, two showed DRD symptoms, five had PD phenotype, and four were asymptomatic. The only patient with SPR mutation clinically displayed symptoms of dystonia and parkinsonism. DaTscan® revealed degeneration of the nigroestriatal pathway in two PD patients. Furthermore, phenotypic variability of the same mutation was observed in two families.
ConclusionsDRD associated mutations have a heterogeneous clinical expression GCH1 gene mutations can be manifested with PD phenotype along with the degeneration of the nigroestiatal pathway. Therefore, GCH1 gene testing might be useful in cases of PD suggestive of genetic origin or with family history of dystonia.
La distonía que responde a la dopa (DRD) engloba a un grupo de distonías primarias clínica y genéticamente heterogéneas que se caracterizan por una excelente respuesta al tratamiento con levodopa. La mayoría de estas entidades se deben a un defecto en la vía de la síntesis de la dopamina1. Entre ellas, la DYT/PARK-GCH1 (clásicamente conocida como DYT5a o enfermedad de Segawa), es la más frecuente. Se produce por mutaciones en el gen GCH1 localizado en el brazo largo del cromosoma 14. El gen GCH1 codifica la enzima guanosín trifosfato ciclohidrolasa 1 (GTP-CH-1), limitante en el primer paso de la síntesis de tetrahidrobiopterina, cofactor esencial para la producción de dopamina en las neuronas nigroestriatales (fig. 1). El déficit de esta enzima provoca una gran depleción de la dopamina nigroestriatal2. Su herencia es autosómica dominante con penetrancia incompleta (30-87%)3. En conjunto, las mutaciones del gen GCH1 suponen el 71-87% de casos de DRD4,5. La DYT/PARK-TH (antes conocida como DYT5b) y la DYT/PARK-SPR son formas de DRD autosómica recesiva y resultan del déficit de las enzimas hidroxilasa de tirosina y reductasa de sepiapterina, respectivamente2 (fig. 1). Otras mutaciones no relacionadas directamente con la vía de la síntesis de la dopamina también pueden ser causantes de DRD. Entre ellas, destacan por su frecuencia las del gen PARK2, que pueden presentarse inicialmente como distonía sin parkinsonismo y en las que la aparición de discinesias inducidas por levodopa es muy característica6. Recientemente, también se ha descrito un caso de DRD en un paciente con una mutación del gen LRRK27.

El fenotipo clásico de la DRD es el de inicio en la infancia o adolescencia como distonía focal de miembros inferiores, con posterior progresión a una distonía generalizada con fluctuación diurna de los síntomas1,2. Sin embargo, las manifestaciones clínicas de la DRD son heterogéneas. El desarrollo de parkinsonismo es frecuente, motivo por el que esta entidad se engloba dentro de las distonías combinadas con otros trastornos del movimiento8. Cada vez con más frecuencia se describen casos de mutaciones en el gen GCH1 que comienzan en la edad adulta con parkinsonismo, en ausencia de distonía, indistinguibles clínicamente de la enfermedad de Parkinson (EP) idiopática. En estas formas se ha demostrado la degeneración de la vía nigroestriada, a diferencia de las formas que se inician con distonía9. La variabilidad fenotípica de las mutaciones asociadas a DRD no solo se ha descrito en individuos no emparentados, sino que diferentes estudios han puesto de manifiesto también la variabilidad fenotípica dentro de una misma familia con la misma mutación10.
El objetivo del presente estudio es describir el fenotipo clínico de pacientes con mutaciones en los genes GCH1 y SPR en nuestra unidad.
Pacientes y métodosEstudio retrospectivo y observacional realizado con un registro consecutivo y prospectivo de pacientes con DRD que fueron evaluados por la Unidad de Trastornos del Movimiento del Hospital Clínico Universitario de Santiago de Compostela. El estudio se realizó de conformidad con la Declaración de Helsinki de la Asociación Médica Mundial y fue aprobado por el Comité de Ética en Investigación de Santiago (código de identificación del proyecto 2017/350).
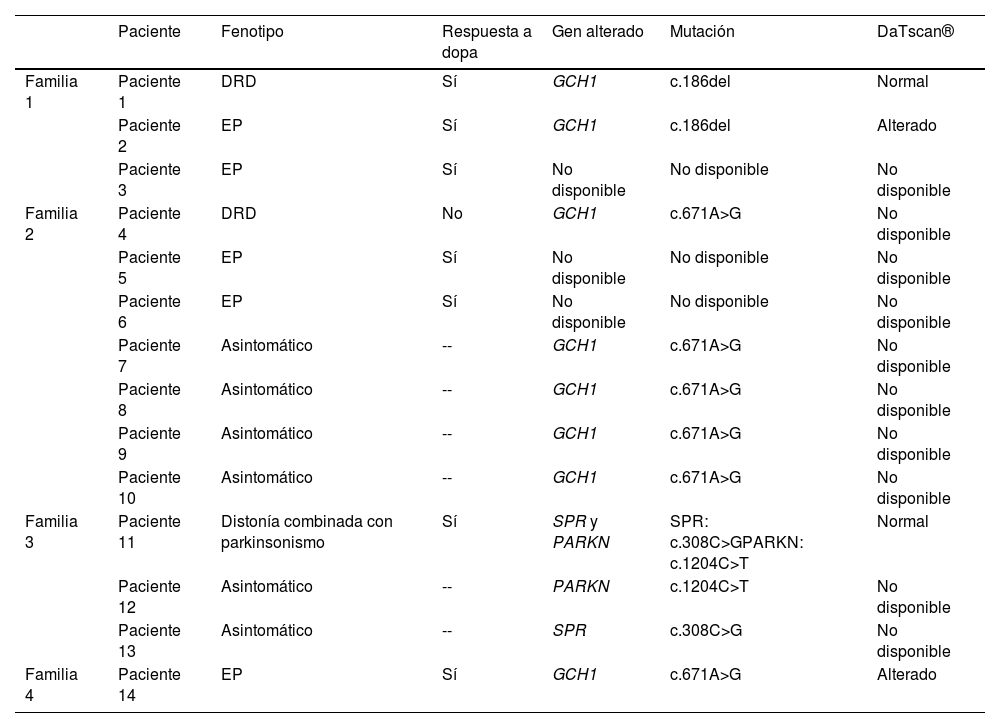
ResultadosSe describen clínica y genéticamente 14 pacientes agrupados en 4 familias que presentan mutaciones asociadas a la DRD (tabla 1).
Características clínicas, genéticas e imagen dopaminérgica de los pacientes incluidos en el estudio
| Paciente | Fenotipo | Respuesta a dopa | Gen alterado | Mutación | DaTscan® | |
|---|---|---|---|---|---|---|
| Familia 1 | Paciente 1 | DRD | Sí | GCH1 | c.186del | Normal |
| Paciente 2 | EP | Sí | GCH1 | c.186del | Alterado | |
| Paciente 3 | EP | Sí | No disponible | No disponible | No disponible | |
| Familia 2 | Paciente 4 | DRD | No | GCH1 | c.671A>G | No disponible |
| Paciente 5 | EP | Sí | No disponible | No disponible | No disponible | |
| Paciente 6 | EP | Sí | No disponible | No disponible | No disponible | |
| Paciente 7 | Asintomático | -- | GCH1 | c.671A>G | No disponible | |
| Paciente 8 | Asintomático | -- | GCH1 | c.671A>G | No disponible | |
| Paciente 9 | Asintomático | -- | GCH1 | c.671A>G | No disponible | |
| Paciente 10 | Asintomático | -- | GCH1 | c.671A>G | No disponible | |
| Familia 3 | Paciente 11 | Distonía combinada con parkinsonismo | Sí | SPR y PARKN | SPR: c.308C>GPARKN: c.1204C>T | Normal |
| Paciente 12 | Asintomático | -- | PARKN | c.1204C>T | No disponible | |
| Paciente 13 | Asintomático | -- | SPR | c.308C>G | No disponible | |
| Familia 4 | Paciente 14 | EP | Sí | GCH1 | c.671A>G | Alterado |
El caso índice (paciente 1) es una mujer que a la edad de 19 años consultó por postura distónica en el pie izquierdo desencadenada con el ejercicio. El estudio genético mostró una mutación en heterocigosis en el gen GCH1 (c.186del). Los síntomas mejoraron con el tratamiento con levodopa y actualmente se mantiene asintomática con dosis de 100mg/día. Recientemente, se realizó un DaTscan® que resultó normal (fig. 2).

Su padre (paciente 2), en seguimiento previo en nuestro centro, estaba diagnosticado de EP desde los 50 años. Se realizó un DaTscan® al inicio de su seguimiento que mostró una hipocaptación putaminal izquierda. Cumple todos los criterios clínicos de la enfermedad, y actualmente se encuentra en una fase avanzada, debido a fluctuaciones motoras y discinesias de pico de dosis. Ha rechazado la cirugía de estimulación cerebral. El estudio del gen GCH1 detectó la misma mutación que en el caso índice.
El abuelo paterno de la paciente 1 (paciente 3) también estuvo diagnosticado de EP hasta su fallecimiento con 92 años. Cumplía los criterios clínicos de la enfermedad. No se pudo realizar estudio genético ni estudio de la vía nigroestriada.
Familia 2El caso índice (paciente 4) es una mujer de 40 años que comenzó en la adolescencia con distonía focal cervical. Atendiendo a sus antecedentes familiares (ver a continuación) se realizó un estudio genético que mostró una mutación en el exón 6 del gen GCH1 en heterocigosis (c.671A>G). Se hizo una prueba con levodopa hasta una dosis de 300mg/día que no fue efectiva. Presenta un buen control con infiltraciones periódicas de toxina botulínica.
Su padre (paciente 5) estaba diagnosticado de EP de predominio rígido-acinético desde los 50 años, por lo que recibía tratamiento con agonistas dopaminérgicos y levodopa. El estudio genético del gen GCH1 resultó positivo. Su abuelo paterno (paciente 6), en la década de 1950, se diagnosticó de EP con marcada dificultad para caminar e inclinación de tronco. Nunca recibió tratamiento ni se realizó estudio genético.
Dos hermanos varones de la paciente 4 (pacientes 7 y 8), su hija (paciente 9) y una sobrina (paciente 10, hija del paciente 7) son portadores asintomáticos de la misma mutación del gen GCH1.
Familia 3El caso índice (paciente 11) es una mujer de 14 años que fue valorada por distonía con inversión del pie derecho. En los meses siguientes asoció temblor en la extremidad inferior derecha y en la exploración se evidenció rigidez y bradicinesia derecha. La respuesta al tratamiento con levodopa fue positiva. El estudio del gen GCH1 no mostró alteraciones, sin embargo, se detectó una mutación en heterocigosis de significado incierto (c.1204C>T) en el gen PRKN y una mutación en heterocigosis (c.308C>G) en el gen SPR. Los datos actuales sobre la patogenicidad de esta mutación en SPR son contradictorios. Se realizó un DaTscan® que fue normal. Dada la presentación clínica, la respuesta al tratamiento y el resultado del DaTscan®, se estableció el diagnóstico de probable DRD.
A los 5 años de seguimiento la paciente presentó un empeoramiento clínico con fluctuaciones motoras. Por ello se decidió realizar una cirugía de estimulación cerebral profunda (ECP) palidal bilateral en septiembre de 2022, con muy buena respuesta clínica.
Se detectó en su padre (paciente 12) la misma mutación de PRKN y en su madre (paciente 13) la mutación c.308C>G de SPR. Ambos están asintomáticos.
Familia 4El caso índice (paciente 14) es un varón que consultó a los 32 años por torpeza y temblor en el brazo derecho. Se realizó un DaTscan® que mostró una hipocaptación putaminal izquierda (fig. 3). El estudio inicial de genes relacionados con EP juvenil no detectó ninguna alteración. Se realizó posteriormente estudio del gen GCH1 que mostró una mutación en heterocigosis (c.671A>G). La respuesta a levodopa fue favorable y muestra estabilidad clínica. El paciente no tenía antecedentes familiares de EP o distonía. No se realizó estudio genético de sus familiares.
Discusión
Las manifestaciones clínicas de las mutaciones asociadas a DRD son heterogéneas. La presentación clásica es la de inicio en la infancia con distonía focal de extremidades inferiores que puede progresar a distonía generalizada2,11. En los casos analizados encontramos como ejemplo de este fenotipo clásico a la paciente 1, con una mutación en heterocigosis del gen GCH1. Sin embargo, la afectación focal puede involucrar a otras zonas corporales11. La paciente 4 es un ejemplo de una localización menos habitual de la DYT/PARK-GCH1, con distonía focal cervical. En este sentido, la afectación cervical en las mutaciones del gen GCH1 se ha descrito en un 12% de los casos11.
Además del fenotipo clásico, la asociación de distonía con parkinsonismo en la DRD es frecuente12. La paciente 11 es un ejemplo de fenotipo combinado, que se inició en su adolescencia con distonía de miembro inferior y parkinsonismo derecho y que respondió adecuadamente al tratamiento con levodopa. El estudio genético de la paciente mostró una mutación en heterocigosis en el gen PRKN y en el gen SPR. Aunque la DYT/PARK-SPR suele tener un patrón de herencia recesivo11,12, se ha comunicado un caso en heterocigosis13, si bien la mutación descrita es diferente a la de nuestro caso. Nos planteamos si la presencia conjunta de la mutación en heterocigosis de PRKN podría tener un efecto sinérgico en este caso.
No solo la asociación con parkinsonismo es común, sino que con cada vez más frecuencia se describen casos de DRD relacionados con mutaciones en el gen GCH1 que comienzan con parkinsonismo en ausencia de distonía. Esta forma de presentación puede ser clínicamente indistinguible de la EP idiopática1,9,11. Durante mucho tiempo, se consideró que el parkinsonismo que se observaba en la DRD era resultado de una reducción significativa de la actividad de GTP-CH-19. En este sentido, es relevante destacar que, inicialmente, se requería la demostración de la integridad de la vía nigroestriada para el diagnóstico diferencial de la DRD con la EP1,2. Sin embargo, actualmente se ha descrito la degeneración de la vía dopaminérgica nigroestriatal en pacientes con mutaciones en el gen GCH1 que comienzan con parkinsonismo9,14–16 y se ha llegado a detectar una prevalencia de mutaciones en el gen GCH1 de hasta el 1,9% en pacientes con EP idiopática en un estudio de realizado en Japón1. Este conocimiento hace que actualmente la demostración de la integridad de la vía nigroestriada no sea un requisito para el diagnóstico de DRD. En nuestra serie de casos, al menos los casos 2 y 14, y presumiblemente los casos 3 y 6, constituyen ejemplos de mutaciones en el gen GCH1 que se manifiestan como EP. Estos casos se manifestaron precozmente, en concordancia con lo descrito en otros estudios1,11 y con otras formas genéticas de la EP17.
El mecanismo por el que las mutaciones del gen GCH1 pueden producir la degeneración de la vía nigroestriada no se conoce con exactitud. Dado que el déficit de GTP-CH-1 provoca una reducción de la dopamina nigroestriatal, se ha sugerido que niveles bajos de esta molécula podrían actuar como factor predisponente precipitante, al hacer al circuito más vulnerable a otros factores genéticos o ambientales9,18. El estudio mediante pruebas de neuroimagen funcional de la vía nigroestriatal dopaminérgica aporta, por tanto, una información relevante en estos casos. En nuestra muestra se realizó estudio de DaTscan® en 4 pacientes con mutaciones en GCH1. En aquellos con fenotipo clásico (paciente 1) y combinado distonía-parkinsonismo (paciente 11) no se observaron alteraciones, pero sí en aquellos que cumplían los criterios clínicos de la EP (pacientes 2 y 14).
Dentro de la heterogeneidad clínica de la DRD, cabe destacar las diferentes formas de manifestación de las mutaciones del gen GCH1 dentro de una misma familia. Ya en 1990, en un estudio se examinó a 106 miembros de una familia afecta de DRD y se observaron sujetos que padecían distintos grados de distonía generalizada, distonía focal y parkinsonismo19. Otro estudio describió la expresión de una misma mutación de GCH1 en forma de DRD, parkinsonismo e incluso de un fenotipo similar a la atrofia multisistema dentro de una misma familia20. Más recientemente, se estudió el fenotipo clínico y la integridad de la vía nigroestriada en 12 miembros de una familia portadores de una misma mutación en GCH1. Los sujetos que mostraron fenotipo DRD mantenían la integridad de la vía dopaminérgica mientras que aquéllos con fenotipo de EP tenían la vía alterada10. En nuestra serie de casos se observa también la heterogeneidad fenotípica de las mutaciones en GCH1 dentro de una misma familia. En la familia 1, mientras que el caso índice presenta fenotipo de DRD, las 2 generaciones anteriores presentaban EP. En la familia 2, el caso índice presenta distonía focal cervical, su padre y su abuelo paterno estaban diagnosticados de EP y su hija, 2 hermanos varones y su sobrina son portadores asintomáticos, al menos por el momento, de la misma mutación de GCH1.
No está claro por qué una misma mutación puede dar lugar a fenotipos diferentes, pero quizás esta variabilidad fenotípica intrafamiliar sea otro dato a favor de la influencia de otros factores ambientales o genéticos en el desarrollo clínico de la alteración de GCH1, como hemos apuntado anteriormente. Además, la penetrancia de las mutaciones en el gen GCH1 es incompleta, entre un 30 y un 87%3. Esto puede explicar la ausencia de síntomas en los pacientes 7, 8, 9 y 10, portadores de una mutación en heterocigosis de GCH1.
Entre las características principales de la DRD destaca la respuesta excelente y sostenida al tratamiento con levodopa1. Sin embargo, la tasa de respuesta a levodopa se ha cifrado en un 86%11, por lo que la ausencia de respuesta no permite descartar el diagnóstico. En nuestra serie, la paciente 2 constituye un caso de distonía focal que no respondió a levodopa pero que presentó mejoría clínica con infiltraciones periódicas de toxina botulínica. Además de la toxina botulínica existen otras alternativas terapéuticas que pueden mejorar la calidad de vida de estos pacientes. La ECP se ha propuesto como tratamiento eficaz en casos de DRD21,22. En este sentido, la paciente 11, que mostró una buena respuesta inicial a levodopa, no toleró aumentos posteriores de dosis por efectos adversos periféricos. En esta paciente se realizó ECP palidal bilateral con una respuesta excelente.
ConclusionesLas mutaciones asociadas a la DRD presentan una expresión clínica heterogénea. En concreto, las mutaciones en el gen GCH1 constituyen un factor de riesgo para la EP. En los casos en los que se presentan como EP se ha demostrado la degeneración de la vía nigroestriada y la edad de inicio suele ser más temprana, acorde a otras formas de EP genéticas.
En este sentido, el estudio del gen GCH1 puede ser de utilidad en aquellos casos de EP sugestivos de origen genético o con antecedentes familiares de distonía.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

