




La ganglionopatía autonómica autoinmune (GAA) es un trastorno inmunomediado adquirido1. Fue descrito originalmente en 1974 como una pandisautonomía pura y se le consideró una variante del síndrome de Guillain-Barré2. En 1994 se identificó en estos pacientes presencia de anticuerpos específicos contra la subunidad alfa-3 del receptor de acetilcolina (ACh) ganglionar3. La mayoría de los casos son agudos o subagudos y son precedidos por una infección respiratoria2. Presentamos un caso con disautonomía de progresión lenta, con estudio de fibras delgadas.
Un varón de 43 años sin antecedentes relevantes, que alrededor de un año antes de la primera evaluación comenzó a presentar en forma lenta y progresiva síntomas de intolerancia ortostática con mareos, visión borrosa al ponerse de pie y debilidad en las piernas al caminar. Estos síntomas se aliviaban en decúbito. Posteriormente, comenzó a presentar síncopes frecuentes y fue evaluado por un cardiólogo, quien descartó una cardiopatía; la prueba de Tilt test mostró presencia de hipotensión ortostática (HO) y fue enviado para estudio autonómico. Evaluación: el paciente refirió presencia de nicturia, disfunción eréctil, sequedad oral y ocular, ausencia de sudor con el ejercicio e intolerancia a la luz. Examen neurológico: función cognitiva normal. Pares craneanos: midriasis fija bilateral, motilidad ocular normal. v al xii par normales. Examen motor y sensitivo normales. Pruebas autonómicas: compromiso simpático cardiovascular, cardiovagal, sudomotor y pupilar (tabla 1). La analítica no mostró anemia ni leucocitosis, velocidad de sedimentación globular, glucemia, creatinina, niveles de vitamina B12 y ácido fólico normales. Virus de la inmunodeficiencia humana negativo, gammaglobulinas séricas normales; inmunología: anticuerpos Ro/La y antinucleares negativos. Pruebas de imágenes sin lesiones tumorales. Conducción nerviosa motora y sensitiva normales. Biopsia de piel: cuantificación normal de la densidad de fibras amielínicas en la epidermis y dermis (glándulas sudoríparas).
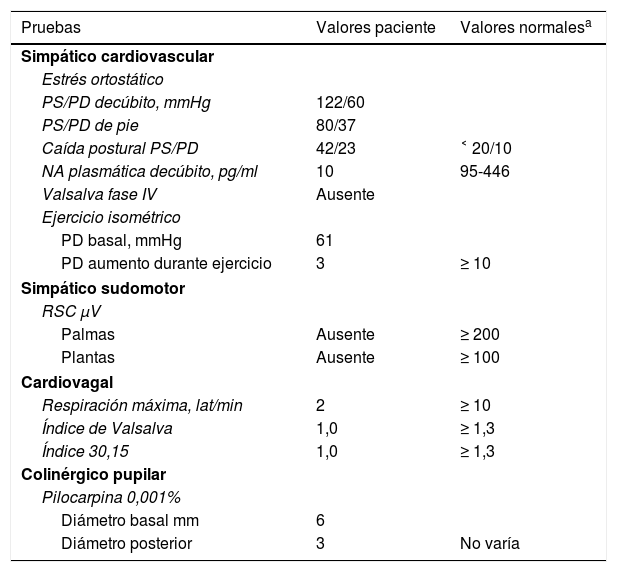
Resultados del estudio de la función autonómica
| Pruebas | Valores paciente | Valores normalesa |
|---|---|---|
| Simpático cardiovascular | ||
| Estrés ortostático | ||
| PS/PD decúbito, mmHg | 122/60 | |
| PS/PD de pie | 80/37 | |
| Caída postural PS/PD | 42/23 | ˂ 20/10 |
| NA plasmática decúbito, pg/ml | 10 | 95-446 |
| Valsalva fase IV | Ausente | |
| Ejercicio isométrico | ||
| PD basal, mmHg | 61 | |
| PD aumento durante ejercicio | 3 | ≥ 10 |
| Simpático sudomotor | ||
| RSC μV | ||
| Palmas | Ausente | ≥ 200 |
| Plantas | Ausente | ≥ 100 |
| Cardiovagal | ||
| Respiración máxima, lat/min | 2 | ≥ 10 |
| Índice de Valsalva | 1,0 | ≥ 1,3 |
| Índice 30,15 | 1,0 | ≥ 1,3 |
| Colinérgico pupilar | ||
| Pilocarpina 0,001% | ||
| Diámetro basal mm | 6 | |
| Diámetro posterior | 3 | No varía |
RSC: respuesta simpática cutánea; NA: noradrenalina plasmática; PD: presión arterial diastólica; PS: presión arterial sistólica.
El diagnóstico diferencial de esta disautonomía simpática y parasimpática de evolución gradual sin presencia de compromiso motor ni sensitivo incluyó: el síndrome de Sjögren, que puede presentar una disautonomía asociada a un compromiso de fibras sensitivas gruesas o delgadas4, pero en este caso hubo ausencia de anticuerpos específicos y de neuropatía sensitiva. También la amiloidosis puede presentarse por disautonomía asociada a una polineuropatía periférica axonal5. Este paciente no presentaba compromiso cardiaco, renal ni de nervios periféricos. Otro diagnóstico fue una disautonomía paraneoplásica, que se presenta en forma aguda o subaguda como un cuadro de seudoobstrucción intestinal aislada o asociada a encefalitis o a ganglionopatía sensitiva6, este paciente no presentó compromiso encefálico ni de nervios periféricos, además la exploración sistémica toracoabdominal y prostática fue negativa. También se consideró el fallo autonómico puro (FAP), el cual se manifiesta por un compromiso simpático y parasimpático sin déficit motor ni sensitivo lentamente progresivo, debido a un proceso neurodegenerativo crónico (sinucleinopatía)7.
El paciente presentó una midriasis fija bilateral debida a denervación colinérgica pupilar; este hallazgo es frecuente en casos de GAA con títulos elevados de anticuerpos y es raro en pacientes con FAP8, por este motivo se solicitó el estudio de autoanticuerpos antirreceptor de ACh ganglionar. La determinación de autoanticuerpos anti receptor de ACh ganglionar, subunidad alfa fue positiva, concentración mayor a 3,6 nmol/l (nivel normal ˂ 0,05 nmol/l, Universidad Southwestern, TX, EE. UU.). La HO se trató con midodrina y fludrocortisona. Recibió terapia inmunosupresora con prednisona y azatioprina; en los meses siguientes el paciente tuvo una moderada mejoría de la intolerancia ortostática, con presencia ocasional de síncopes. Seguimiento por 4años no mostró reaparición de síncopes ni presencia de nuevos síntomas.
Las manifestaciones importantes de la GAA son la HO, estreñimiento, síntomas colinérgicos prominentes pupilares, urinarios, ojos y boca seca e intolerancia al calor por anhidrosis1,2. Los autoanticuerpos bloquean la trasmisión ganglionar provocando la disautonomía1. En el tratamiento de la GAA aguda se utiliza la inmunoglobulina por vía intravenosa o plasmaféresis, a largo plazo se usa prednisona, micofenolato, azatioprina, rituximab solos o en combinación1,2. En el presente caso de progresión lenta se usó una combinación de prednisona y azatioprina con beneficio terapéutico. Pacientes con el cuadro clínico de GAA pueden tener niveles negativos de autoanticuerpos anti receptor de ACh y presentan un compromiso simpático predominante sin signos pupilares y responden mejor a corticoides9.
En este caso, el estudio morfológico mostró una inervación conservada de la glándula sudorípara, lo que apoya un defecto en la trasmisión sin lesión neuronal ni axonal (figura 1). Sin embargo, un bloqueo prolongado de la trasmisión ganglionar podría causar perdida de fibras delgadas10. La GAA con progresión lenta puede ser diagnosticada inicialmente como FAP11, por este motivo es importante identificar estos casos que pueden mejorar con la terapia inmunosupresora a diferencia de la FAP, que un trastorno neurodegenerativo.
, la flecha tipo guión muestra fibras epidérmicas con densidad normal (10,1 fibras por mm, lo que es –0,9 desviaciones del promedio para su edad y sexo). A la derecha (marcador fluorescente DAPI), la flecha blanca muestra un conducto sebáceo inervado en forma normal.")
Biopsia de piel: a la izquierda (marcador pan neuronal PGP9.5), la flecha tipo guión muestra fibras epidérmicas con densidad normal (10,1 fibras por mm, lo que es –0,9 desviaciones del promedio para su edad y sexo). A la derecha (marcador fluorescente DAPI), la flecha blanca muestra un conducto sebáceo inervado en forma normal.
Agradecemos al Dr. Steven Vernino de Texas University Southwestern Medical Center, por sus consejos y por el estudio de los anticuerpos.

