





Los anticuerpos contra un complejo proteico que incluye a los canales de potasio dependientes de voltaje (CKVD) se han descrito en pacientes con encefalitis límbica, hiperexcitabilidad del nervio periférico, síndrome de Morvan, así como en un creciente grupo de síndromes neurológicos.
DesarrolloEn este artículo revisamos los síndromes asociados a anticuerpos contra proteínas relacionadas con los CKVD y los 2 antígenos principales de este complejo, las proteínas leucine rich glioma inactivated protein 1 (LGI1) y contactin-associated protein-like 2 (Caspr2). Así mismo describimos los problemas conceptuales y las implicaciones diagnósticas de la descripción de anticuerpos contra CKVD diferentes de LGI1 y Caspr2.
Aunque inicialmente se consideró que existían anticuerpos dirigidos contra CKVD, recientemente se ha identificado que, en la mayor parte de los casos, los antígenos son una proteína neuronal secretada denominada LGI1, involucrada en el control de la excitabilidad sináptica, y la proteína Caspr2, localizada en la superficie neuronal de varias regiones cerebrales y en la región yuxtaparanodal de axones mielinizados. Mientras que los anticuerpos contra LGI1 se asocian preferentemente a un cuadro clásico de encefalitis límbica, los anticuerpos contra Caspr2 muestran un espectro clínico más amplio, incluyendo el síndrome de Morvan, la hiperexcitabilidad del nervio periférico o neuromiotonía, o una encefalitis límbica o difusa. Existen además casos descritos de pacientes con anticuerpos contra el complejo CKVD que no tienen anticuerpos contra LGI1 o Caspr2. En estos casos, la identidad y la localización de los antígenos es desconocida, la asociación sindrómica inespecífica y la respuesta al tratamiento, incierta.
ConclusionesEl descubrimiento de los antígenos LG1 y Caspr2ha permitido delimitar clínica y molecularmente el amplio grupo de síndromes previamente atribuidos a anticuerpos contra CKVD. Frente a la literatura que describe la presencia de anticuerpos contra CKVD diferentes a LGI1 y Caspr2, proponemos un algoritmo práctico para el diagnóstico y el tratamiento de estos pacientes.
Antibodies against a protein complex that includes voltage-gated potassium channels (VGKC) have been reported in patients with limbic encephalitis, peripheral nerve hyperexcitability, Morvan's syndrome, and a large variety of neurological syndromes.
Review summaryIn this article, a review is presented of the syndromes associated with antibodies against VGKC-related proteins and the main antigens of this protein complex, the proteins LGI1 (leucine rich glioma inactivated protein 1) and Caspr2 (contactin-associated protein-like 2). The conceptual problems and clinical implications of the description of antibodies against VGKC-related proteins other than LGI1 and Caspr2 are also discussed.
Although initial studies indicated the occurrence of antibodies against VGKC, recent investigations have shown that the main antigens are a neuronal secreted protein known as LGI1 which modulates synaptic excitability, and a protein called Caspr2 located on the cell surface and processes of neurons of different brain regions, and at the juxtaparanodal region of myelinated axons. While antibodies against LGI1 preferentially associate with classical limbic encephalitis, antibodies against Caspr2 associate with a wider spectrum of symptoms, including Morvan's syndrome, peripheral nerve hyperexcitability or neuromyotonia, and limbic or more extensive encephalitis. In addition there are reports of patients with antibodies against VGKC-related proteins that are different from LGI1 or Caspr2. In these cases, the identity and location of the antigens are unknown, the syndrome association is not specific, and the response to treatment uncertain.
ConclusionsThe discovery of antigens such as LGI1 and Caspr2 has resulted in a clinical and molecular definition of the broad group of diseases previously attributed to antibodies against VGKC. Considering the literature that describes the presence of antibodies against VGKC other than LGI1 and Caspr2 proteins, we propose a practical algorithm for the diagnosis and treatment of these patients.
Los anticuerpos anticanales de potasio dependientes de voltaje (CKVD) se han identificado en el contexto de un amplio espectro de síndromes neurológicos con afectación del sistema nervioso central y periférico tanto en adultos1 como en niños2. Inicialmente, se pensaba que estos anticuerpos estaban dirigidos contra epítopos del propio canal, pero en los últimos años se ha descrito que la mayoría de ellos se unen a la proteína 1 inactivada del glioma rica en leucina (leucine rich glioma inactivated protein 1 [LGI1])3 y a la proteína símil 2 asociada a contactina (contactin-associated protein-like 2 [Caspr2])3,4. Además, recientemente ha emergido un grupo de pacientes con anticuerpos atribuidos a proteínas del complejo CKVD pero negativos para Caspr2 y LGI15,6.
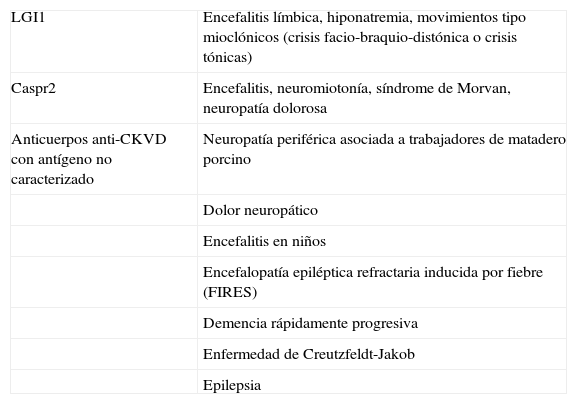
La presencia de anticuerpos contra LGI1 ocurre en el contexto de encefalitis límbica3, mientras que los anticuerpos contra Caspr2 pueden asociarse a encefalitis3,4, hiperexcitabilidad del nervio periférico (neuromiotonía adquirida o síndrome de Isaacs)7, o a la combinación de ambas (síndrome de Morvan)3-6. Se trata, en ambos casos, de proteínas bien caracterizadas, cuya alteración sustenta fisiopatológicamente los cuadros clínicos de las correspondientes respuestas autoinmunes. Por el contrario, la naturaleza del antígeno de los pacientes que presentan anticuerpos contra el complejo CKVD pero negativos para LGI1 y Caspr2 es incierta8. Además, el perfil clínico de los pacientes con estos anticuerpos es heterogéneo y progresivamente creciente. Por todo ello, no es de extrañar que se planteen dudas sobre el significado clínico y patogénico de estos anticuerpos (tabla 1). De esta manera, y con el objetivo de clarificar el conocimiento actual del espectro clínico-patológico y delimitar el marco sindrómico asociado a estos anticuerpos, presentamos este trabajo de revisión.
Espectro clínico asociado a la detección de anticuerpos anti-CKVD
| LGI1 | Encefalitis límbica, hiponatremia, movimientos tipo mioclónicos (crisis facio-braquio-distónica o crisis tónicas) |
| Caspr2 | Encefalitis, neuromiotonía, síndrome de Morvan, neuropatía dolorosa |
| Anticuerpos anti-CKVD con antígeno no caracterizado | Neuropatía periférica asociada a trabajadores de matadero porcino |
| Dolor neuropático | |
| Encefalitis en niños | |
| Encefalopatía epiléptica refractaria inducida por fiebre (FIRES) | |
| Demencia rápidamente progresiva | |
| Enfermedad de Creutzfeldt-Jakob | |
| Epilepsia |
El término anticuerpos anti-CKVD se empleó para definir anticuerpos detectados por técnica de radioinmunoanálisis (RIA) contra el complejo de proteínas que incluyen las subunidades Kv1.1 y Kv1.2 de la familia Shaker de los CKVD. La positividad de este test viene definida por la unión del radiotrazador α-I125-dendrotoxina a antígenos que forman parte de un conglomerado proteico que precipita junto con las proteínas de los CKVD. Sin embargo, por las características intrínsecas de la técnica, no es posible conocer la identidad de los antígenos, ni siquiera si son neuronales, axonales o sinápticos.
Paralelamente, mediante técnicas de inmunofluorescencia con cultivos de neuronas de hipocampo, se determinó que los antígenos atribuidos a CKVD se localizaban en la superficie neuronal y su identidad se conoció al precipitarlos directamente con los anticuerpos de pacientes y secuenciarlos. Una vez conocida su identidad, LGI13 o Caspr24, se desarrollaron técnicas diagnósticas altamente específicas con células que expresan estos antígenos (cell-based assay [CBA]) (fig. 1).
 y un paciente con anticuerpos contra Caspr2 (D y E) muestra intensa reactividad con el neurópilo que no se observa con el LCR de un paciente control (G y H). La localización de los antígenos (LGI1 y Caspr2) en la superficie neuronal se demuestra en inmunocitoquímica con cultivos de neuronas hipocampales de rata incubados con los LCR de los mismos pacientes (C y F). El panel I muestra la ausencia de reactividad del LCR de un paciente control con cultivos de neuronas. La identidad de los antígenos (LGI1, Caspr2) se demostró en células HEK293 transfectadas (CBA) que expresaban estas proteínas (no se muestra en esta figura).")
Reactividad del LCR de pacientes con anticuerpos contra LGI1 o Caspr2 en cerebro de rata y cultivos de neuronas. La inmunohistoquímica en cerebro de rata utilizando el LCR de un paciente con anticuerpos contra LGI1 (A y B) y un paciente con anticuerpos contra Caspr2 (D y E) muestra intensa reactividad con el neurópilo que no se observa con el LCR de un paciente control (G y H). La localización de los antígenos (LGI1 y Caspr2) en la superficie neuronal se demuestra en inmunocitoquímica con cultivos de neuronas hipocampales de rata incubados con los LCR de los mismos pacientes (C y F). El panel I muestra la ausencia de reactividad del LCR de un paciente control con cultivos de neuronas. La identidad de los antígenos (LGI1, Caspr2) se demostró en células HEK293 transfectadas (CBA) que expresaban estas proteínas (no se muestra en esta figura).
A pesar de estos avances, algunos investigadores mantienen la técnica de RIA para detectar anticuerpos contra el complejo CKVD, describiendo que en el 39-68% de los casos5,6 se encuentran anticuerpos anti-CKVD sin presencia de anticuerpos LGI1 o Caspr2. El problema creado por estos estudios, en los que no se especifica la naturaleza de estos antígenos mediante el empleo de técnicas suplementarias, se analiza más adelante (Sección anticuerpos contra el complejo CKVD diferentes de LGI1 y Caspr2).
LGI1 es una proteína neuronal secretada que interactúa a nivel presináptico con ADAM 23 y a nivel postsináptico con ADAM 22, organizando un complejo proteico transináptico que se ha relacionado en humanos con casos de epilepsia9. Otros componentes de este complejo incluyen las subunidades Kv1.1 y Kv1.2 a nivel presináptico y el receptor ácido α-amino-3-hidroxi-5 metil-4-isoxazolpropiónico (AMPA) a nivel postsináptico. Mutaciones en el gen que codifica LGI1 se han asociado a la epilepsia autosómica dominante del lóbulo temporal lateral o a epilepsia parcial autosómica dominante con síntomas auditivos10,11, una forma de epilepsia hereditaria caracterizada por crisis parciales con alucinaciones visuales o auditivas. Esta mutación en modelos animales se ha correlacionado con un aumento de la excitabilidad neuronal, que se ha atribuido a la disminución de la actividad del receptor AMPA en neuronas inhibitorias9 y al aumento de la liberación de glutamato12. Se ha propuesto que esta hiperexcitabilidad contribuye a los problemas de memoria y epilepsia que manifiestan pacientes con anticuerpos anti-LGI13.
Caspr2 es una proteína axonal transmembrana de la superfamilia de las neurexinas, que se une a contactina 2 y colabora en la organización de canal de potasio Kv1 en la región yuxtaparanodal, donde parece que desempeña un papel en el correcto funcionamiento de los axones mielinizados13. Además, está presente también a nivel del hipocampo y del cerebelo14. Las mutaciones y los polimorfismos en el gen que codifica Caspr2 (CNTNAP2) se han detectado en el contexto de pacientes psiquiátricos y con epilepsia resistente, así como en casos de hiperexcitabilidad del nervio periférico14-17. Estos hechos sustentan fisiopatológicamente el espectro clínico asociado a la presencia de anticuerpos contra Caspr2.
Espectro clínico asociado a la detección de anticuerpos contra LGI1Los anticuerpos anti-LGI1 se han asociado con un cuadro de encefalitis límbica clásica caracterizada por la pérdida de memoria a corto plazo y crisis epilépticas. Epidemiológicamente, en la serie publicada por Lai et al.3, parece que existe predominancia en varones con edades comprendidas en torno a la sexta década de la vida, aunque el rango de edad abarca desde casos infantiles hasta los 80 años. Clínicamente, se caracteriza por un cuadro de encefalitis límbica de inicio subagudo, con sintomatología y orden de instauración variables, que acaba desembocando en un déficit amnésico asociado frecuentemente a un trastorno de conducta del sueño REM18, y en un alto porcentaje de casos a crisis epilépticas. Semiológicamente, estas últimas pueden manifestarse como crisis generalizadas, temporales mesiales y hasta en un 40% de los casos como crisis tónicas19. La frecuencia de las crisis tónicas en este tipo de casos ha llevado a pensar que puedan constituir una señal de alarma en el diagnóstico diferencial con otros casos de encefalitis límbicas19. Característicamente, se manifiestan por movimientos bruscos, espasmódicos, asimétricos y breves, de carácter tónico predominantemente, aunque pueden asociar un componente distónico que afecta principalmente a la región orofacial y los miembros superiores, motivo por el cual algunos autores las han denominado crisis faciobraquiales distónicas20. Sin embargo, 3 son los aspectos que pueden llevar al clínico a confusión con respecto a esta denominación: 1) los movimientos no se restringen a los miembros superiores y la región orofacial en todos los casos; 2) la existencia de una correlación electroencefalográfica de los mismos con una respuesta decremental en el rango de frecuencias β característica, aunque no patognomónica, de las crisis tónicas, y 3) la respuesta al tratamiento antiepiléptico en algunos casos20. Estas 2 últimas premisas apuntan a que el mecanismo patogénico subyacente más probable sea epileptógeno frente a que se trate de un trastorno del movimiento.
En relación con los estudios complementarios, es característica la asociación a hiponatremia en un 60% de los casos3,6. Los estudios de rutina del líquido cefalorraquídeo (LCR) suelen ser normales, aunque los pacientes pueden presentar una linfocitosis moderada y un aumento de la concentración de proteínas. En la RM, el 84% de los pacientes tienen un aumento de señal en secuencias potenciadas en T2, a nivel temporal medial, uni o bilateralmente.
La asociación a tumores es infrecuente, en cuyo caso lo más habitual es que se trate de un timoma. En el 70-80% de pacientes los síntomas responden al tratamiento con esteroides, inmunoglobulinas por vía intravenosa o recambio plasmático. Sin embargo, en muchos pacientes persisten déficits de memoria que les dificultan volver a su trabajo habitual. Las recurrencias de encefalitis son infrecuentes.
Espectro clínico asociado a la detección de anticuerpos CASPR2Los anticuerpos anti-Caspr2 se asocian a síntomas que pueden afectar tanto al sistema nervioso central en forma de encefalitis, como al sistema nervioso periférico en forma de hiperexcitabilidad del nervio periférico, o su combinación (síndrome de Morvan). Es difícil determinar la frecuencia de presentación de estas formas y si alguna de ellas prevalece sobre las demás, ya que el número de pacientes incluidos en las diferentes series descritas hasta el momento es limitado.
La encefalitis asociada a la presencia de anticuerpos anti-Caspr2 es un cuadro que se presenta en torno a los 60 años, aunque se han descrito casos entre los 19 y los 80 años, con una aparente predominancia en varones4,21. Clínicamente, se manifiesta como una encefalitis multifocal o difusa, a veces límbica, que en la mayor parte de los casos se asocia a hiperexcitabilidad del nervio periférico en forma de neuromiotonía estricta o en formas incompletas con calambres y fasciculaciones. Esta neuromiotonía puede preceder al desarrollo del cuadro encefalítico en un periodo variable. No es rara la presencia de crisis epilépticas de semiología diversa, incluida la presencia de crisis tónicas4,21.
En los casos de síndrome de Morvan, se ha descrito una mayor prevalencia de trastornos del sueño, siendo el insomnio lo más habitual4,21, que en algunas ocasiones puede llegar a niveles de agrypnia excitata22. Además, hasta en un 62% de los casos presentan dolor neuropático asociado frecuentemente a arreflexia e hipoestesia en guante y calcetín. La disfunción autonómica en forma de hiperhidrosis o inestabilidad cardiovascular puede estar presente hasta en el 93% de los casos21. Por otro lado, se ha detectado a pacientes que presentan síntomas bulbares, fasciculaciones y debilidad en los miembros inferiores asociados a la presencia de anticuerpos contra el receptor de acetil colina o anticuerpos anticinasa específica muscular, planteando un diagnóstico diferencial con enfermedad de motoneurona4.
El análisis del LCR está alterado hasta en el 25% de los casos, en forma generalmente de pleocitosis o proteinorraquia, pudiendo presentar adicionalmente síntesis de bandas oligoclonales4. En algunos, pacientes la RM muestra un aumento de señal en secuencias potenciadas en T2 a nivel temporal medial21, aunque esta suele ser normal en el contexto del síndrome de Morvan4,5,21.
Con respecto a la asociación tumoral, los datos disponibles en la literatura son dispares. Así, mientras algunos grupos identifican tumor en torno al 20-41%5,21 de los casos, predominantemente timomas y en el contexto de un síndrome de Morvan, otros grupos apuntan a que se trata de una posibilidad más remota4. En relación con el tratamiento, parece que, en líneas generales, el pronóstico es bueno, excepto en los casos en los que se asocian a tumor4,5,21, pero no es infrecuente la recidiva.
A pesar de que clásicamente se ha asociado la presencia de anticuerpos contra el complejo de CKVD con neuromiotonía adquirida aislada23,24, estudios más recientes apuntan a que esta relación es probablemente más infrecuente de lo que se pensaba25.
Síndromes y síntomas atribuidos a anticuerpos contra el complejo canales de potasio dependientes de voltaje: antígenos desconocidos, valor clínico inciertoA pesar de que la detección de los anticuerpos LGI1 y Caspr2ha contribuido a clarificar el amplio espectro clínico de síndromes asociados a la detección de anticuerpos contra el complejo CKVD, existe un grupo heterogéneo de síndromes clínicos en los que se ha detectado la presencia de estos anticuerpos y ausencia de anticuerpos LGI1 y Caspr2. Este amplio conjunto sindrómico abarca tanto cuadros restringidos exclusivamente al sistema nervioso central (encefalopatía epiléptica refractaria inducida por fiebre, epilepsia, encefalitis infantiles, demencia rápidamente progresiva y enfermedad de Creutzfeldt-Jakob), como a síndromes del sistema nervioso periférico (neuropatía, dolor neuropático)2,26-29. En todos estos cuadros, los antígenos son desconocidos; ni siquiera se sabe si la localización de estos antígenos es intra o extracelular. Además, la respuesta al tratamiento inmunosupresor en los casos descritos es muy variable. Todo esto lleva a plantearse el valor diagnóstico y patogénico de estos anticuerpos. El escaso valor diagnóstico de estos anticuerpos quedó reflejado en un estudio reciente en el que los autores clasificaban el síndrome como autoinmune o no en función de la respuesta a la inmunoterapia, sin tener en cuenta si los pacientes tenían anticuerpos CKVD30. Por tanto, más investigaciones son necesarias para poder dilucidar el significado clínico de estos anticuerpos.
Manejo diagnóstico y terapéutico práctico frente a pacientes con anticuerpos atribuidos a canales de potasio dependientes de voltajeAtendiendo a lo descrito, ante la sospecha clínica de un síndrome neurológico autoinmune asociado a la presencia de anticuerpos contra el complejo proteico que incluye los CKVD, proponemos el algoritmo indicado en la figura 2. En esta figura, los pasos a seguir se recomiendan mediante líneas sólidas. La línea discontinua indica que cualquiera que sea el resultado con RIA, se va a necesitar la confirmación diagnóstica con otras pruebas más específicas, como inmunocitoquímica con células que expresan LGI1 o Caspr2 y, si esta es negativa, pruebas adicionales con cultivos de neuronas para determinar si los antígenos están en la superficie celular o no. La identificación de reactividad del suero y/o LCR con la superficie de neuronas vivas indica con alta probabilidad que el síndrome va a responder a la inmunoterapia y, por lo tanto, esta debe iniciarse independientemente de la identidad del antígeno. Por otra parte, si el suero y/o el LCR no reaccionan con la superficie de neuronas vivas pero reaccionan con secciones de cerebro de rata, los antígenos son intracelulares u onconeuronales y, por lo tanto, se recomienda el análisis de anticuerpos paraneoplásicos clásicos, y si estos son positivos, la búsqueda y el tratamiento del tumor. Los síndromes asociados a anticuerpos contra antígenos intracelulares o paraneoplásicos suelen responder menos a la inmunoterapia que los asociados a anticuerpos contra antígenos de la superficie neuronal.

Algoritmo para el diagnóstico de pacientes con anticuerpos contra el complejo proteico que incluye los CKVD.
*Síndromes clínicos descritos en la tabla 1.
**NMDAR, AMPAR, GABAbR, DPPX, GlyR, otros.
Las inmunoterapias de primera línea más frecuentemente utilizadas para LGI1, Caspr2 y otros antígenos de superficie son esteroides, inmunoglobulinas por vía intravenosa y recambio plasmático de forma individual o combinada. A efectos prácticos, la combinación más frecuente es esteroides con inmunoglobulinas por vía intravenosa. Si no hay respuesta a estas inmunoterapias, la estrategia seguida con la encefalitis por anticuerpos contra el receptor NMDA, es decir rituximab y/o ciclofosfamida, es la más razonable31.
Conflicto de interesesEl Dr. Dalmau tiene la patente para el uso de los tests diagnósticos para los anticuerpos Ma2 y receptor NMDA, y ha solicitado la patente para el uso de los anticuerpos contra los receptors GABAa y GABAb como tests diagnósticos.
FinanciaciónEl Dr. Dalmau tiene financiación del Instituto Nacional de la Salud de Estados Unidos (NIH, RO1NS077851), Fundació la Marató TV3, y Instituto Carlos III (FIS, PI11/01780).

