




El síndrome FOXG1 es una encefalopatía epiléptica-discinética descrito inicialmente como una variante del síndrome de Rett (SR, MIM: 613454); el cual es un desorden del neurodesarrollo causado en la mayoría de los casos por mutaciones en el gen MECP2 y afecta exclusivamente a mujeres. El SR se caracteriza por regresión de los hitos del desarrollo entre los 6 a 18 meses, movimientos estereotipados de las manos, microcefalia, convulsiones y retardo mental. Es una enfermedad con un espectro fenotípico amplio y presenta un componente genético heterogéneo, el cual se ha asociado a microdeleciones y translocaciones en diferentes genes como CDKL5/STK9, NTNG1, y recientemente el factor de trascripción cerebral FOXG1 se ha visto involucrado como parte del espectro genotípico de este síndrome. El FOXG1 (locus: 14q12) presenta un gran rol en el desarrollo cerebral1, ya que regula el desarrollo embrionario temprano del telencéfalo en estado fetal y adulto2. Presentamos a un paciente con una mutación heterocigota en este gen, con el objetivo de plantear las alteraciones en este gen como causa del síndrome FOXG1 y no como una variante del SR.
Describimos a un paciente de origen colombiano, masculino de 3 años de edad al momento de la evaluación, producto del segundo embarazo de padres consanguíneos, embarazo sin complicaciones. Gestación de 37 semanas, parto por cesárea, peso al nacer de 2.680g (p10-50), talla al nacer 47cm (p10-50), perímetro cefálico 35,5cm (p50-90). Periodo perinatal sin complicaciones, a los 3 meses de edad es valorado por neuropediatría donde encuentran fontanela anterior puntiforme, tono ligeramente aumentado. Por estos hallazgos se solicita tomografía axial computarizada de cráneo con reconstrucción tridimensional, la cual evidenció opérculos abiertos, fosa temporal en las puntas amplias, reconstrucción tridimensional sin sinostosis, microcráneo con occipucio plano, perímetro cefálico−2 desviaciones estándar (DE). Adicionalmente el paciente cursó con un retraso en el desarrollo psicomotor y enfermedad por reflujo gastroesofágico, por lo cual es remitido a oftalmología y genética clínica. En oftalmología pediátrica encuentran fijación parcial binocular, movimientos repetitivos de la cabeza, comportamiento visual no esperado para la edad, asociado a retraso global del desarrollo.
A la evaluación clínica a los 3 años se encuentra paciente de peso 8,7kg (−2,25 DE), estatura 82cm (−0,53 DE), peso/talla (−2,75 DE), IMC 12,9kg/mt2 (−2,91 DE) perímetro cefálico 40cm (−2 DE), microcráneo con occipucio plano, sutura metópica prominente, frente prominente, estrabismo convergentes, puente nasal bajo, filtrum plano, orejas en anteversión, protrusión lingual, escroto en shalt. Por los hallazgos clínicos y del desarrollo se realizan estudios metabólicos y pruebas para mucopolisacaridosis, los cuales fueron negativos. Cariotipo con bandeo G mostrando un complemento cromosómico reportado como normal (46, XY) e hibridación genómica comparativa con microarreglos, normal con zonas de homocigosidad, se realizó una secuenciación del exoma en trío, utilizando kit TruSight One de Illumina, bajo la plataforma Next Seq 500. Se llevó a cabo un análisis selectivo sobre los genes relacionados con el diagnóstico clínico del paciente, detectando una variante en el gen FOXG1 (NM_005249.4): c.1107:1108insG, p.Glu371GlyfsTer84 en estado heterocigoto, la cual es una inserción de una guanina que da lugar a una parada prematura en la transcripción y por tanto una proteína truncada; dicha variante no ha sido reportada previamente en la literatura, sin embargo se han descrito mutaciones que afectan la transcripción en el mismo aminoácido c.460dupG, p.e154GfsX301 y se han reportado como patogénicas, ya que resulta en pérdida de los 3 dominios de unión a las proteínas3. La variante candidata se confirmó mediante secuenciación Sanger y análisis de esta en los padres, comprobando el origen de novo. Se concluye que la sumatoria de hallazgos clínicos: pobre crecimiento y microcefalia posnatal, pobre contacto visual e interacción social, estrabismo convergente, protrusión lingual, enfermedad por reflujo gastroesofágico, pie plano, hipotonía neonatal, irritabilidad neonatal, retraso del desarrollo motor, apraxia, movimiento estereotipados, ausencia de lenguaje, patrón de sueño anormal, electroencefalograma, resonancia magnética normal y el patrón de herencia aislado, genera una correlación genotipo-fenotipo con el SR atípico causado por mutaciones en el FOXG1.
La mutación del FOXG1 es extremadamente rara y se encuentra en el 1-2% de los pacientes con sospecha de síndrome del espectro autista, con una frecuencia del 1,5% en las mujeres4. El gen FOXG1 es un represor transcripcional, con una alta expresión en la región ventricular y neuroepitelial del telencéfalo, estructuras visuales y tejido testicular5, el cual promueve la proliferación de las células progenitoras y a su vez es un supresor prematuro de la neurogénesis y diferenciación neuronal6. A través del reclutamiento de proteínas represoras transcripcionales Groucho y JARID1C que participan en la metilación de la histona 3 en el residuo de lisina 4, lo que genera silenciamiento de la cromatina7. Análisis moleculares han demostrado que este gen comparte vías metabólicas con el MeCP2 durante el desarrollo neuronal y presenta una superposición parcial de su perfil de expresión posnatal en la corteza cerebral y las regiones subnucleares8.
El gen FOXG1 es el gen candidato del síndrome por microdeleción 14q12 como enfermedad monogénica. En el caso presentado se encuentra un paciente con retraso del desarrollo psicomotor; pese a esto logra sentarse y caminar, explicado por la haploinsuficiencia del FOXG1 que genera espectros clínicos menos severos donde una mayor proporción de los pacientes observados en las series alcanzan los hitos del desarrollo como caminar (3/8 pacientes)8. Algunos estudios han concluido que las variantes heterocigotas del FOXG1 no necesariamente resultan en malformaciones cerebrales9, como se puede evidenciar en el caso presentado.
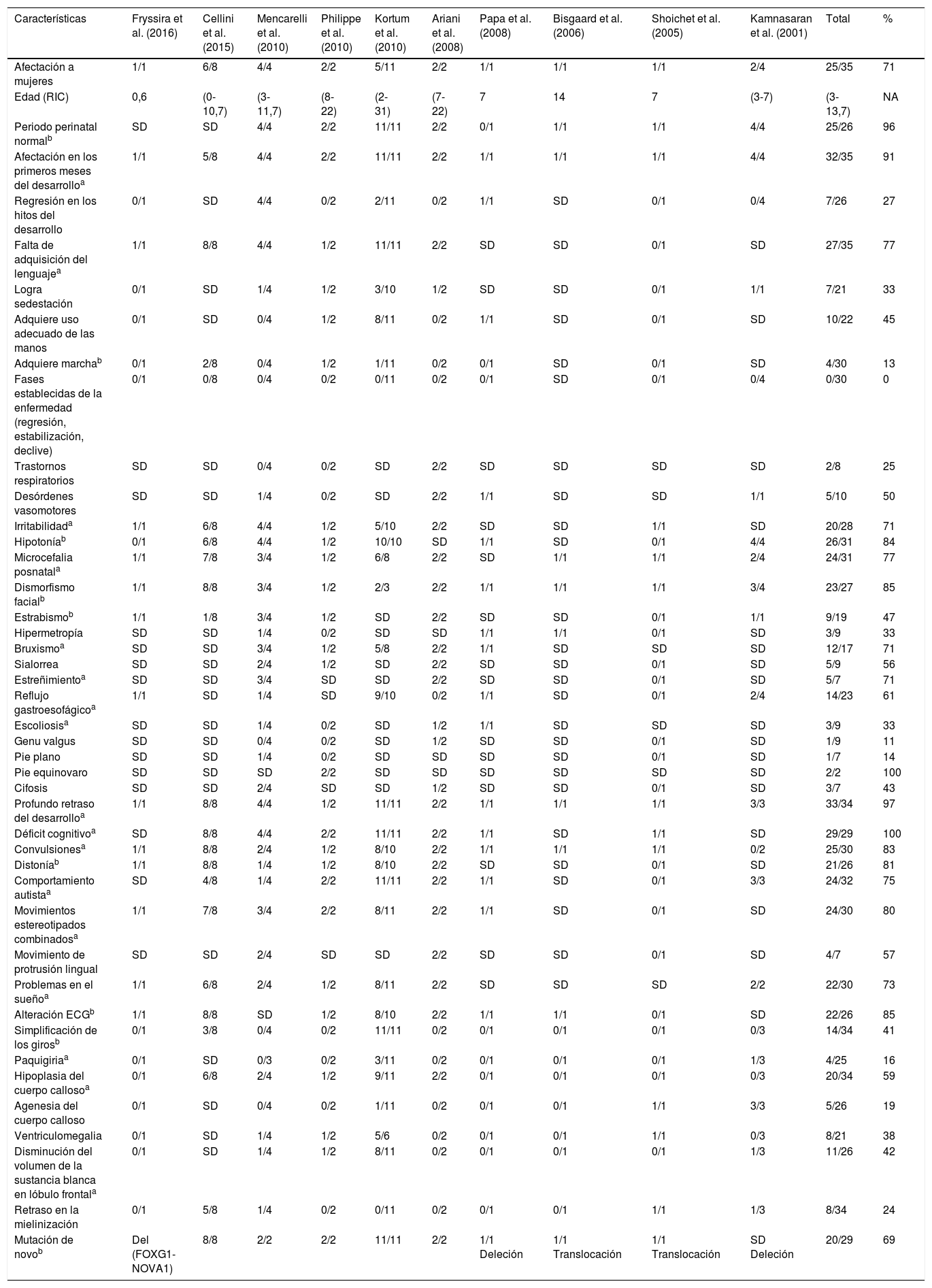
En el SR atípico se han identificado diferentes genes asociados como CDKL5/STK9, NTNG1 y FOXG1, pese a esto las mutaciones en este último gen generan espectros clínicos más variados que difieren ampliamente de las características clínicas del SR. Por esta razón algunos autores han planteado este como una entidad que difiere clínica y genotípicamente del SR y presenta características clínicas distintivas como las discinesias hipercinéticas10. Se analizaron 35 pacientes con mutaciones puntuales, deleciones, translocaciones FOXG1, en todos los casos de origen de novo, previamente reportados en la literatura, que muestran un fenotipo dependiente de una gran variabilidad genotípica. Este síndrome afecta ambos sexos, en el 69% de los casos mujeres, en gran proporción de los casos hay ausencia de regresión de los hitos del desarrollo y fases establecidas de la enfermedad, típico de SR, al contrario, afectación tempranamente en el periodo neonatal. En la tabla 1 se reconocen características del síndrome FOXG1 previamente planteadas por Kortum et al.10 y sugerimos adicionar otras características debido a que se presentan en más del 75% de los casos reportados, ampliando y estableciendo mejor el fenotipo de estos pacientes.
Características de los casos reportados en la literatura
| Características | Fryssira et al. (2016) | Cellini et al. (2015) | Mencarelli et al. (2010) | Philippe et al. (2010) | Kortum et al. (2010) | Ariani et al. (2008) | Papa et al. (2008) | Bisgaard et al. (2006) | Shoichet et al. (2005) | Kamnasaran et al. (2001) | Total | % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Afectación a mujeres | 1/1 | 6/8 | 4/4 | 2/2 | 5/11 | 2/2 | 1/1 | 1/1 | 1/1 | 2/4 | 25/35 | 71 |
| Edad (RIC) | 0,6 | (0-10,7) | (3-11,7) | (8-22) | (2-31) | (7-22) | 7 | 14 | 7 | (3-7) | (3-13,7) | NA |
| Periodo perinatal normalb | SD | SD | 4/4 | 2/2 | 11/11 | 2/2 | 0/1 | 1/1 | 1/1 | 4/4 | 25/26 | 96 |
| Afectación en los primeros meses del desarrolloa | 1/1 | 5/8 | 4/4 | 2/2 | 11/11 | 2/2 | 1/1 | 1/1 | 1/1 | 4/4 | 32/35 | 91 |
| Regresión en los hitos del desarrollo | 0/1 | SD | 4/4 | 0/2 | 2/11 | 0/2 | 1/1 | SD | 0/1 | 0/4 | 7/26 | 27 |
| Falta de adquisición del lenguajea | 1/1 | 8/8 | 4/4 | 1/2 | 11/11 | 2/2 | SD | SD | 0/1 | SD | 27/35 | 77 |
| Logra sedestación | 0/1 | SD | 1/4 | 1/2 | 3/10 | 1/2 | SD | SD | 0/1 | 1/1 | 7/21 | 33 |
| Adquiere uso adecuado de las manos | 0/1 | SD | 0/4 | 1/2 | 8/11 | 0/2 | 1/1 | SD | 0/1 | SD | 10/22 | 45 |
| Adquiere marchab | 0/1 | 2/8 | 0/4 | 1/2 | 1/11 | 0/2 | 0/1 | SD | 0/1 | SD | 4/30 | 13 |
| Fases establecidas de la enfermedad (regresión, estabilización, declive) | 0/1 | 0/8 | 0/4 | 0/2 | 0/11 | 0/2 | 0/1 | SD | 0/1 | 0/4 | 0/30 | 0 |
| Trastornos respiratorios | SD | SD | 0/4 | 0/2 | SD | 2/2 | SD | SD | SD | SD | 2/8 | 25 |
| Desórdenes vasomotores | SD | SD | 1/4 | 0/2 | SD | 2/2 | 1/1 | SD | SD | 1/1 | 5/10 | 50 |
| Irritabilidada | 1/1 | 6/8 | 4/4 | 1/2 | 5/10 | 2/2 | SD | SD | 1/1 | SD | 20/28 | 71 |
| Hipotoníab | 0/1 | 6/8 | 4/4 | 1/2 | 10/10 | SD | 1/1 | SD | 0/1 | 4/4 | 26/31 | 84 |
| Microcefalia posnatala | 1/1 | 7/8 | 3/4 | 1/2 | 6/8 | 2/2 | SD | 1/1 | 1/1 | 2/4 | 24/31 | 77 |
| Dismorfismo facialb | 1/1 | 8/8 | 3/4 | 1/2 | 2/3 | 2/2 | 1/1 | 1/1 | 1/1 | 3/4 | 23/27 | 85 |
| Estrabismob | 1/1 | 1/8 | 3/4 | 1/2 | SD | 2/2 | SD | SD | 0/1 | 1/1 | 9/19 | 47 |
| Hipermetropía | SD | SD | 1/4 | 0/2 | SD | SD | 1/1 | 1/1 | 0/1 | SD | 3/9 | 33 |
| Bruxismoa | SD | SD | 3/4 | 1/2 | 5/8 | 2/2 | 1/1 | SD | SD | SD | 12/17 | 71 |
| Sialorrea | SD | SD | 2/4 | 1/2 | SD | 2/2 | SD | SD | 0/1 | SD | 5/9 | 56 |
| Estreñimientoa | SD | SD | 3/4 | SD | SD | 2/2 | SD | SD | 0/1 | SD | 5/7 | 71 |
| Reflujo gastroesofágicoa | 1/1 | SD | 1/4 | SD | 9/10 | 0/2 | 1/1 | SD | 0/1 | 2/4 | 14/23 | 61 |
| Escoliosisa | SD | SD | 1/4 | 0/2 | SD | 1/2 | 1/1 | SD | SD | SD | 3/9 | 33 |
| Genu valgus | SD | SD | 0/4 | 0/2 | SD | 1/2 | SD | SD | 0/1 | SD | 1/9 | 11 |
| Pie plano | SD | SD | 1/4 | 0/2 | SD | SD | SD | SD | 0/1 | SD | 1/7 | 14 |
| Pie equinovaro | SD | SD | SD | 2/2 | SD | SD | SD | SD | SD | SD | 2/2 | 100 |
| Cifosis | SD | SD | 2/4 | SD | SD | 1/2 | SD | SD | 0/1 | SD | 3/7 | 43 |
| Profundo retraso del desarrolloa | 1/1 | 8/8 | 4/4 | 1/2 | 11/11 | 2/2 | 1/1 | 1/1 | 1/1 | 3/3 | 33/34 | 97 |
| Déficit cognitivoa | SD | 8/8 | 4/4 | 2/2 | 11/11 | 2/2 | 1/1 | SD | 1/1 | SD | 29/29 | 100 |
| Convulsionesa | 1/1 | 8/8 | 2/4 | 1/2 | 8/10 | 2/2 | 1/1 | 1/1 | 1/1 | 0/2 | 25/30 | 83 |
| Distoníab | 1/1 | 8/8 | 1/4 | 1/2 | 8/10 | 2/2 | SD | SD | 0/1 | SD | 21/26 | 81 |
| Comportamiento autistaa | SD | 4/8 | 1/4 | 2/2 | 11/11 | 2/2 | 1/1 | SD | 0/1 | 3/3 | 24/32 | 75 |
| Movimientos estereotipados combinadosa | 1/1 | 7/8 | 3/4 | 2/2 | 8/11 | 2/2 | 1/1 | SD | 0/1 | SD | 24/30 | 80 |
| Movimiento de protrusión lingual | SD | SD | 2/4 | SD | SD | 2/2 | SD | SD | 0/1 | SD | 4/7 | 57 |
| Problemas en el sueñoa | 1/1 | 6/8 | 2/4 | 1/2 | 8/11 | 2/2 | SD | SD | SD | 2/2 | 22/30 | 73 |
| Alteración ECGb | 1/1 | 8/8 | SD | 1/2 | 8/10 | 2/2 | 1/1 | 1/1 | 0/1 | SD | 22/26 | 85 |
| Simplificación de los girosb | 0/1 | 3/8 | 0/4 | 0/2 | 11/11 | 0/2 | 0/1 | 0/1 | 0/1 | 0/3 | 14/34 | 41 |
| Paquigiriaa | 0/1 | SD | 0/3 | 0/2 | 3/11 | 0/2 | 0/1 | 0/1 | 0/1 | 1/3 | 4/25 | 16 |
| Hipoplasia del cuerpo callosoa | 0/1 | 6/8 | 2/4 | 1/2 | 9/11 | 2/2 | 0/1 | 0/1 | 0/1 | 0/3 | 20/34 | 59 |
| Agenesia del cuerpo calloso | 0/1 | SD | 0/4 | 0/2 | 1/11 | 0/2 | 0/1 | 0/1 | 1/1 | 3/3 | 5/26 | 19 |
| Ventriculomegalia | 0/1 | SD | 1/4 | 1/2 | 5/6 | 0/2 | 0/1 | 0/1 | 1/1 | 0/3 | 8/21 | 38 |
| Disminución del volumen de la sustancia blanca en lóbulo frontala | 0/1 | SD | 1/4 | 1/2 | 8/11 | 0/2 | 0/1 | 0/1 | 0/1 | 1/3 | 11/26 | 42 |
| Retraso en la mielinización | 0/1 | 5/8 | 1/4 | 0/2 | 0/11 | 0/2 | 0/1 | 0/1 | 1/1 | 1/3 | 8/34 | 24 |
| Mutación de novob | Del (FOXG1-NOVA1) | 8/8 | 2/2 | 2/2 | 11/11 | 2/2 | 1/1 Deleción | 1/1 Translocación | 1/1 Translocación | SD Deleción | 20/29 | 69 |
El síndrome FOXG1 ocurre por mutaciones, al igual que arreglos cromosómicos como deleción o translocación 14q12-q13, es una encefalopatía discinética-epiléptica, con fenotipo identificado10. Al principio fue descrito como una variante del SR, ya que existen muchas características clínicas que se sobreponen con este síndrome, pese a esto existen una gran cantidad de rasgos clínicos que difieren de este. En la tabla 2. Se muestran criterios mayores, menores e imagenológicos de este síndrome, de acuerdo a la frecuencia reportada en las diferentes series de casos.
Criterios mayores, menores e imagenológicos del síndrome FOXG1 según la frecuencia de presentación en los casos reportados en la literatura
| Criterios mayores |
| 1. Mutación, translocación o deleción de novo |
| 2. Déficit cognitivo |
| 3. Periodo perinatal normal, afectación en los primeros meses de vida |
| 4. Microcefalia posnatal |
| 5. Hipotonía |
| 6. Profundo retraso del desarrollo, falta de adquisición del lenguaje |
| 7. Alteraciones en el EEG |
| Criterios menores |
| Irritabilidad, dismorfismo facial (frente prominente, orejas en anteversión, hipertelorismo), estrabismo, bruxismo, estreñimiento, reflujo gastroesofágico, convulsiones, distonías, problemas en el sueño, movimientos estereotipados combinados, comportamiento autista |
| Criterios imagenológicos |
| Paquigiria, simplificación de los giros, hipoplasia del cuerpo calloso, disminución de la sustancia blanca en lóbulo frontal |
La realización de este trabajo, ha sido financiada por la Universidad Icesi.

