








INTRODUCCION
El síndrome de Werner (SW) o progeria del adulto fue descrito en 1904 por el oftalmólogo alemán Carl Wilhelm Otto Werner en su tesis doctoral "sobre cataratas en conexión con esclerodermia", en 4 hermanos con signos de senilidad prematura, lesiones esclerodérmicas y catarata juvenil bilateral1. Existe una progeria infantil, síndrome de Hutchinson-Gilford, con retraso del crecimiento y alopecia ya desde los 2 años. En el SW, el gen responsable es el WRN/SGS1, localizado en el brazo corto del cromosoma 8 (8p12)2, y la teoría más aceptada es una alteración de la proteína de Werner, ADN helicasa-exonucleasa, familia RecQhelicasa3.
Se hereda de forma autosómica recesiva, y es una rara enfermedad con mayor incidencia en grupos con alto indice de consanguinidad, fundamentalmente Japón (1.000 de los 1.300 casos descritos)4 y la Cerdeña italiana. En la clínica predominan la talla baja y el envejecimiento precoz, la piel está atrófica, aparece hiperqueratosis de capa córnea, y frecuentes úlceras en zonas de apoyo, calcificaciones subcutáneas en tendones, atrofia muscular, facies "de pájaro", nariz afilada, pequeña, y poca grasa orbitaria. Así mismo aparecen marcadas arrugas peribucales e hipoplasia mandibular5, voz ronca, por afectación de cuerdas vocales y epiglotis; encanecimiento precoz y alopecia progresiva desde los 20 años6. Por otra parte se producen cataratas bilaterales precoces, posteriores y subcapsulares; desarrollo psicomotor e intelectual normal, aunque en fases avanzadas se describen atrofia cerebral y demencia senil7; baja fertilidad o atrofia gonadal. Se asocia una diabetes tipo 2 en un 50%, con insulinorresistencia y mala respuesta al tratamiento8. Las alteraciones cardiovasculares, por arteriosclerosis avanzada, calcificación de aorta y válvulas cardíacas y arterias de la pierna, con claudicación gangrena del calcáneo, incluso amputaciones por insuficiencia vascular, enfermedad oclusiva femoropoplítea, que precisa by-pass9. También son frecuentes la hipertensión, estenosis valvulares, insuficiencia cardíaca e infartos. Se produce una incidencia de tumores 10 veces mayor que en la población general. Entre los benignos, meningiomas; de los malignos epiteliales, el carcinoma de tiroides. De los no epiteliales, sarcoma de células sinoviales10. Los hallazgos radiológicos más significativos son osteoporosis, calcificación de tejidos blandos, y vasculares en extremidades, y alteraciones neurotróficas en los pies11.
La esperanza media de vida no sobrepasa los 46-50 años, por arterioesclerosis avanzada y tumores. El diagnóstico diferencial debe incluir, además del síndrome de Hutchinson-Gilford, el síndrome de Rothmand, el de Steinner, y fundamentalmente con la esclerodermia.
No existe tratamiento actualmente, por lo que es esencial el diagnóstico precoz para identificar los tumores en los primeros estadios. La cirugía de cataratas, de hallux valgus, injerto de piel para las úlceras, by-pass y el control exhaustivo de los factores de riesgo cardiovascular, diabetes, hipertensión, dislipidemia y tabaquismo, con medidas dietéticas y fármacos ayudan a prevenir o retrasar la arteriosclerosis, mejorando la duración y calidad de vida de estos pacientes.
CASO CLINICO
Se trata de 2 hermanos, de 39 y 45 años actualmente. Valorando sus antecedentes familiares los padres niegan consanguinidad, ni conocer patología similar en sus antepasados; tampoco ellos presentan rasgos fenotípicos sugestivos de SW. Tienen otros 2 hijos, un varón de 43 años (que fue intervenido de un adenocarcinoma vesical a los 40), y una mujer de 35 años (intervenida en la infancia de una comunicación interauricular), ambos igualmente sin rasgos sugerentes de SW.
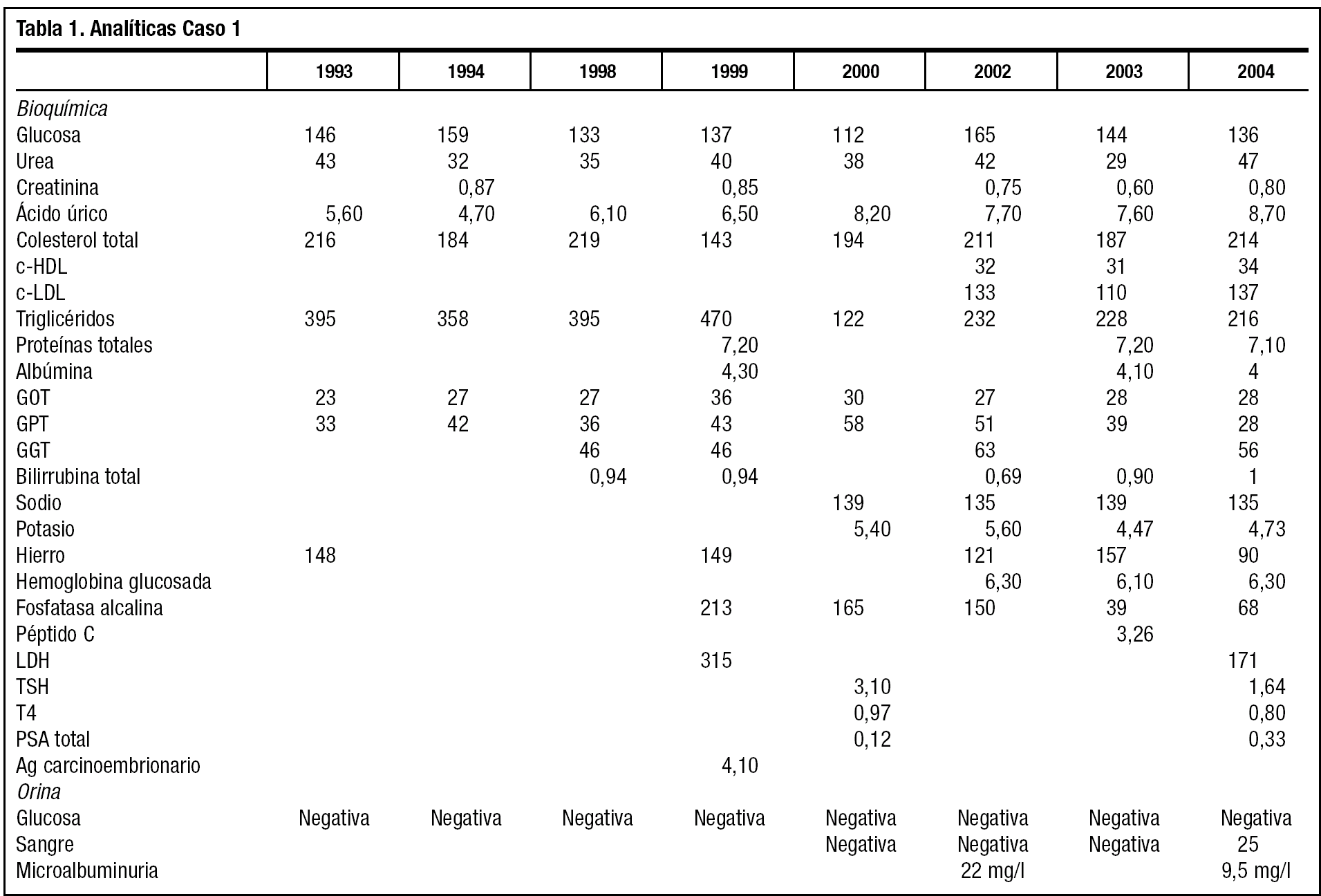
El mayor de los 2 hermanos afectados de SW (caso 1) (fig. 1) es diagnosticado en 1992 de hiperglucemia y dislipidemia por una analítica de un reconocimiento laboral. Acude a consulta de Atención Primaria, y se le abre la historia clínica. Tiene entonces 32 años, una talla de 1,59 m, pesa 51,9 kg, índice de masa corporal 20,52 kg/m2. Fumador de 10 cigarrillos día desde hace más de 10 años, y bebedor de más de 100 g/día de alcohol en fines de semana. Tensión arterial de 140/90 mmHg. Se le aconseja dieta estricta de diabetes e hipolipidemiante, además de abandono del tabaco y restricción severa de alcohol para repetir la analítica. A los 3 meses se obtienen cifras de glucemia 146 mg/dl, colesterol total 216 mg/dl y triglicéridos 395 mg/dl. Ya presenta lesiones atróficas en uñas, y la piel muy seca, acartonada y escamosa. No aparecen déficits visuales ni auditivos. La auscultación cardiopulmonar es normal, aunque refiere palpitaciones ocasionales, tratadas en Servicio de Urgencia de Atención Primaria y Hospital con ansiolíticos y propanolol 10 mg de forma sintomática. El pelo es escaso y encanecido, los labios muy secos y agrietados con marcados pliegues peribucales y periorbitarios, que le confieren un aspecto "de máscara", inexpresivo. Presenta una disfonía severa que ya había sido valorada por el otorrinolaringólogo sin diagnóstico cierto ni tratamiento.
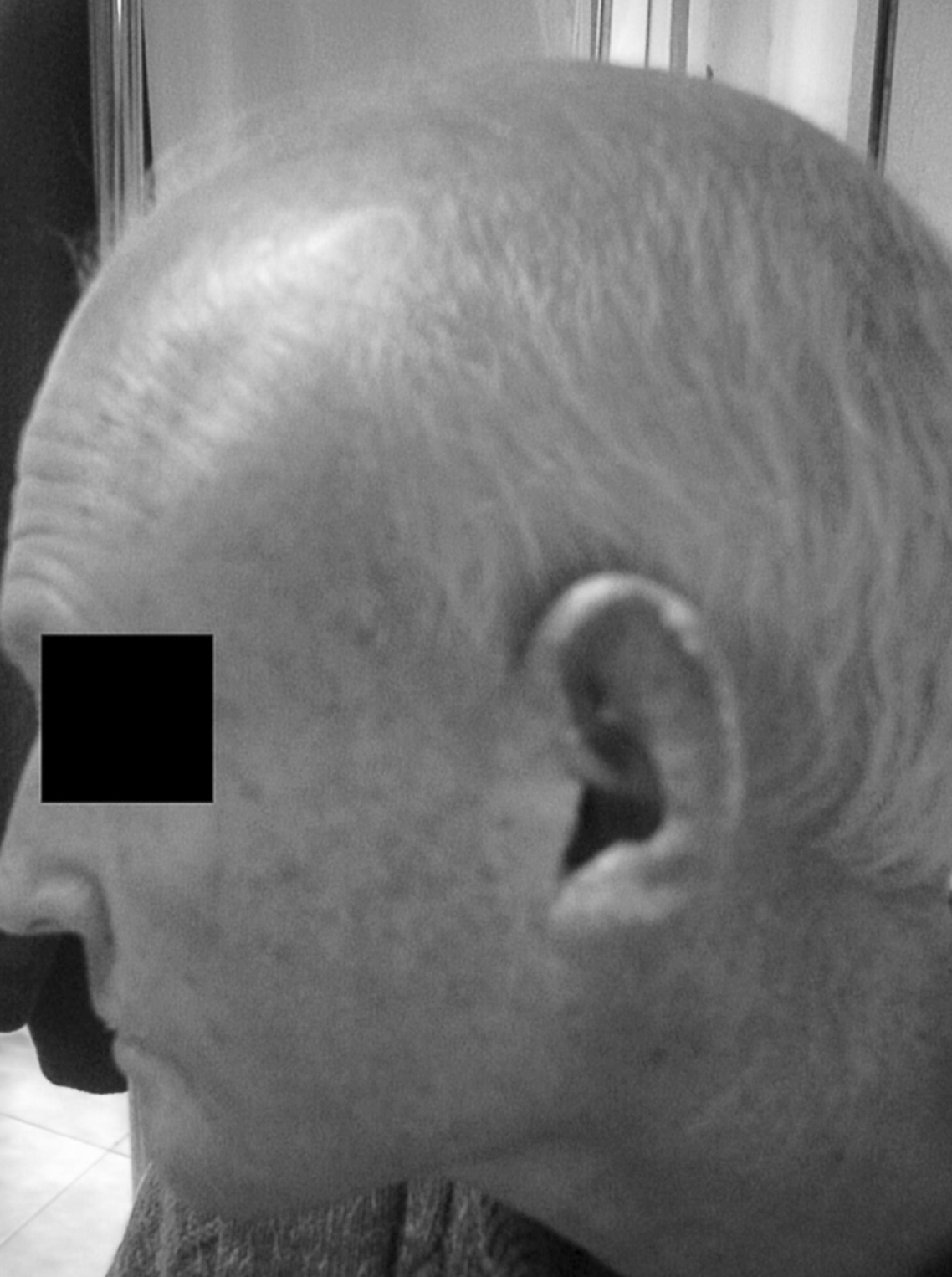
Figura 1. Alopecia marcada.
Por el aspecto descrito, con la sospecha de esclerodermia, se deriva a Reumatología, que añade el diagnóstico de psoriasis y descarta la esclerodermia. Una consulta a Dermatología es la primera indicación diagnóstica del SW. Se deriva posteriormente a Medicina Interna para completar el estudio y cariotipo, que no llegó a realizarse, ya que el paciente deja de acudir a las revisiones que no le aportan tratamiento satisfactorio a sus patologías.
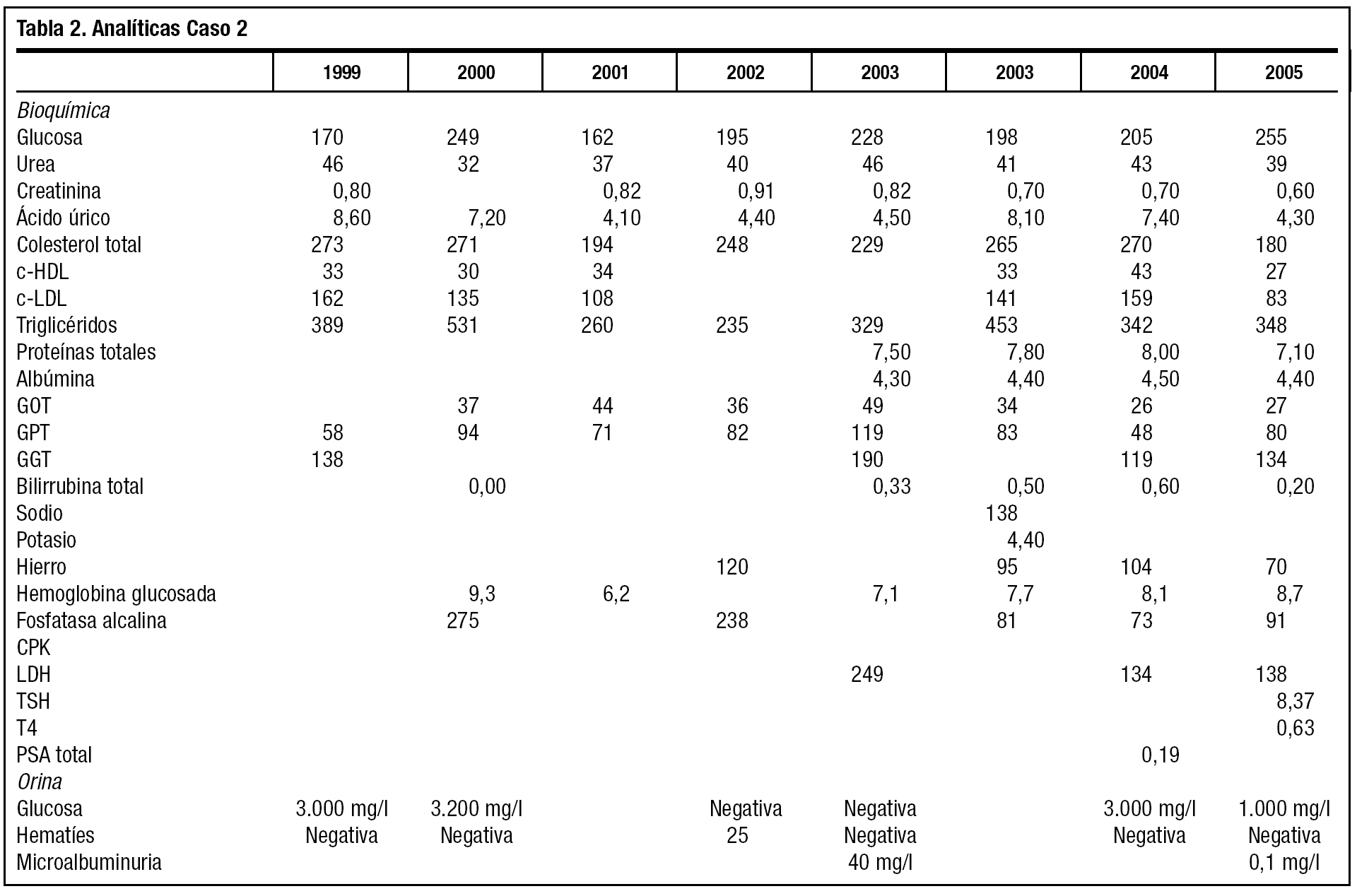
A los 38 años comienza a perder visión y el oftalmólogo confirma cataratas bilaterales, que se intervienen satisfactoriamente con 1 año de diferencia. Había iniciado tratamiento farmacológico con fibratos y antidiabéticos orales, y tras confirmarse la hipertensión, dieta sosa e inhibidores de la enzima de conversión de la angiotensina. El paciente cumple el tratamiento y la dieta muy irregularmente. Trabaja de forma discontinua en tareas agrícolas. Durante los últimos 6 años ha consultado por artralgias y úlceras en los pies y tobillos. Sigue controles de enfermería para su hipertensión arterial, peso, diabetes, además de las curas de sus úlceras, de evolución tórpida. Tiene que utilizar calzado ancho, almohadillado, plantillas y cómodos, para evitar las úlceras. En estos 6 años el pelo se ha encanecido totalmente, y la alopecia es casi completa, salvo en la zona occipital. Además ha sido valorado por Endocrinología, manteniendo el tratamiento con glipizida, simvastatina, alopurinol y enalapril. La evolución bioquímica, como se refleja en la tabla 1, es de aceptable control diabético, a pesar del mal cumplimiento de la dieta y los fármacos (HbA1c < 6,5); la hiperuricemia, hipertrigliceridemia, y la elevación de la GGT en buena parte se explican por la ingesta enólica excesiva. Aparece ya microalbuminuria. Han continuado los episodios de palpitaciones, auscultándose un soplo sistólico II/VI panfocal, y en el electrocardiograma aparece una hipertrofia ventricular izquierda.
A finales de 2004 consulta por una tumoración en cara externa del muslo, de unos 2 cm de diámetro, de reciente aparición. En la radiografía se ve una calcificación subtrocantérea, y como hallazgo casual calcificaciones tendinosas en el pie (fig. 2) y una calcificación vascular severa a nivel femoral (fig. 3). Por todo ello se vuelve a enviar a Medicina Interna, que realiza estudio completo de riesgo cardiovascular. En el ecocardiograma aparece calcificación del anillo mitral, hipertrofia ventricular izquierda e insuficiencia aórtica ligera y en la ecografía abdominal, un hígado graso. La analítica, que incluye PSA, T4, TSH, Ag carcinoembrionario, ácido fólico y B12, es rigurosamente normal. Se confirma la arteriosclerosis precoz y se añade al tratamiento de pentoxifilina y ácido acetilsalicílico 100 mg, y control de sus factores de riesgo por Atención Primaria. El paciente ha dejado de fumar y disminuido la ingesta de alcohol.
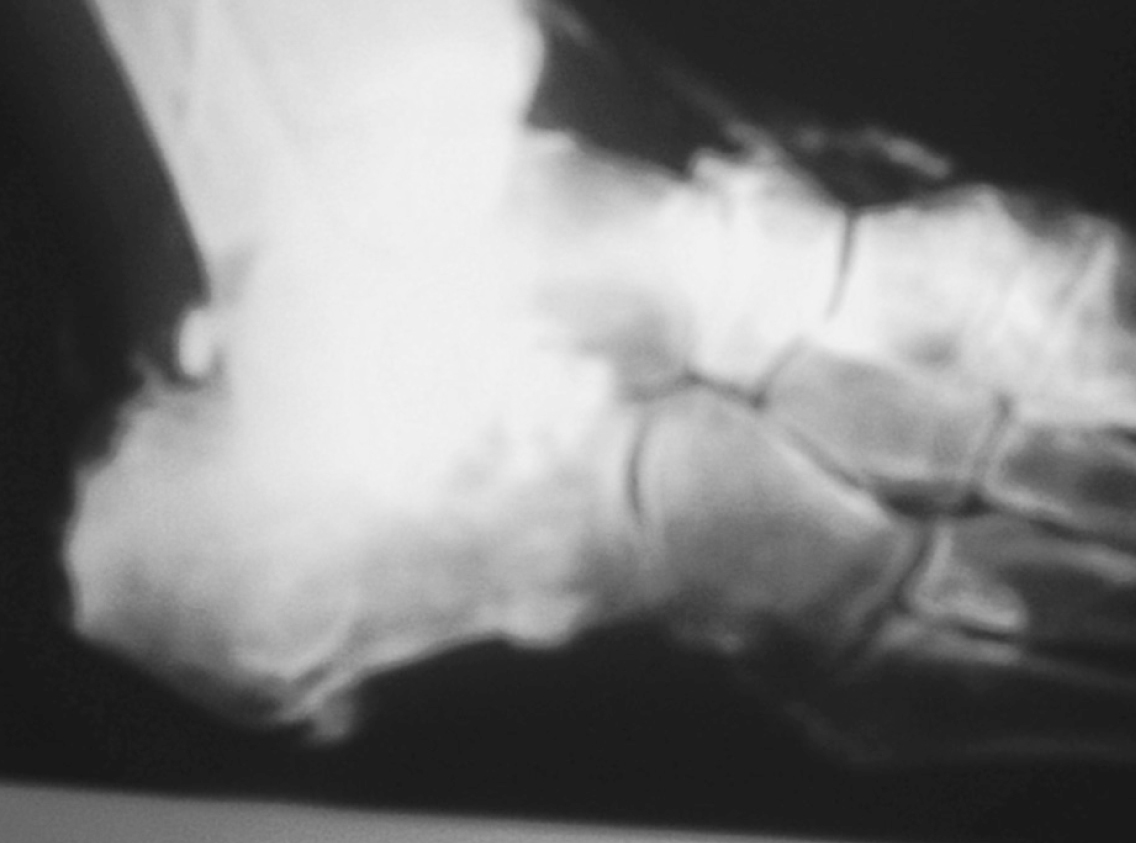
Figura 2. Calcificaciones tendinosas en el pie.
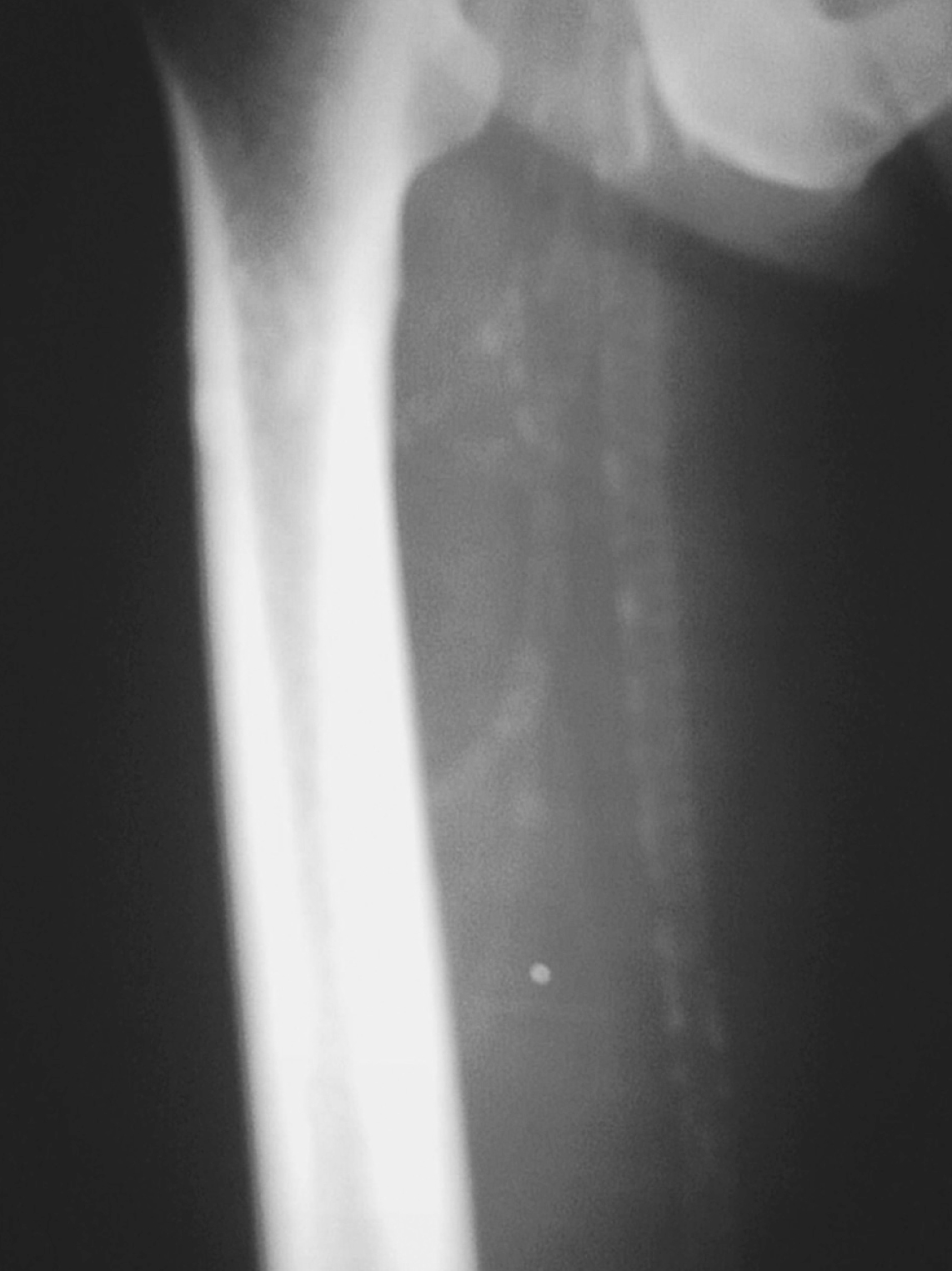
Figura 3. Calcificación severa en la femoral.
El segundo de los hermanos afectados del SW (caso 2) (fig. 4) es 6 años menor, y presenta un fenotipo menos acusado. No existe afonía y mantiene bastante pelo, ya encanecido. La boca es pequeña con pronunciados pliegues peribucales, labios resecos y agrietados; pabellones auriculares apergaminados, con calcificaciones amarillentas o xantomas, y el lóbulo adherido; las manos, con poca elasticidad, aspecto en garra, con atrofia de los músculos de la eminencia tenar; los pies con aspecto similar, atrofia muscular que aumenta los relieves osteotendinosos. Destacan callosidades en talón de Aquiles y cabeza de 1.o y 5.o metatarsianos, con hallux valgus severo en pie izquierdo.
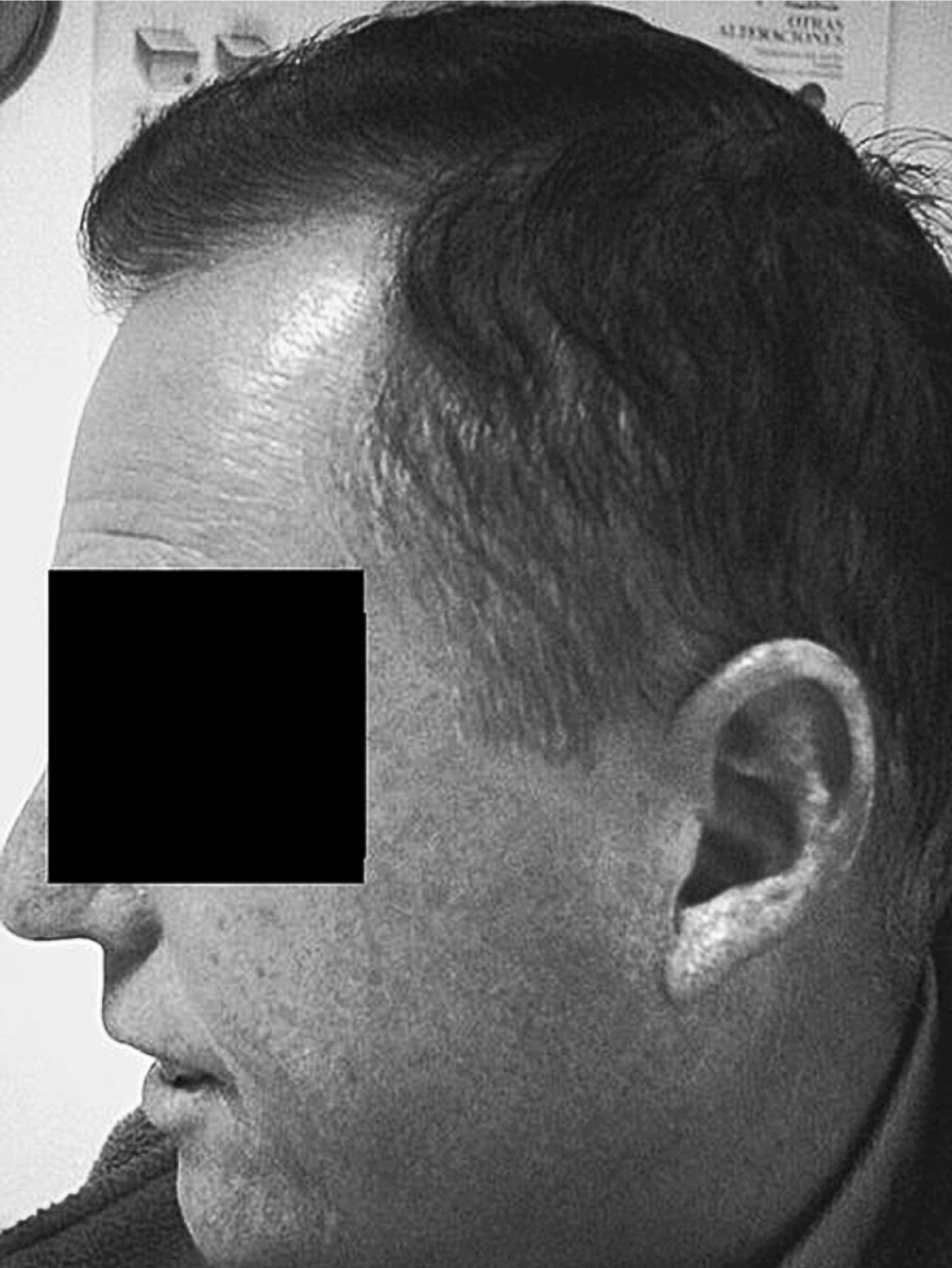
Figura 4. Pliegues peribucales y en pabellón auricular.
El paciente también acude por vez primera a consulta tras hallarse en un análisis de empresa cifras elevadas de glucosa, colesterol, triglicéridos, ácido úrico y transaminasas, en 1999. Fumador de más de 10 cigarrillos/día y bebedor de más de 50 g/día de alcohol en fines de semana. Mide 1,65 m, pesa 60 kg y un índice de masa corporal de 22,58 kg/m2, tensión arterial de 135/90 mmHg. Se pauta dieta sosa, de diabetes y dislipidemia, y al repetir la analítica confirma los diagnósticos de diabetes tipo 2, dislipidemia e hiperuricemia. Se inicia tratamiento con dieta, antidiabéticos orales, fibratos y alopurinol, y se pasa a control de enfermería de su tensión, peso y glucemia.
A los 37 años presenta déficit de agudeza visual y es remitido a Oftalmología e intervenido de cataratas bilaterales a los 38 años. Comienza a presentar úlceras en maleolo peroneo, que tratadas con antisépticos, hidrocoloides y vendajes, tardan más de 2 meses en cicatrizar. Tras presentar una balanitis candidiásica consulta a Urología para intervención de fimosis (¿craurosis?) con postoperatorio sin complicaciones. Dermatología confirma el diagnóstico de SW, ya que seguía la evolución de su hermano desde hacía años.
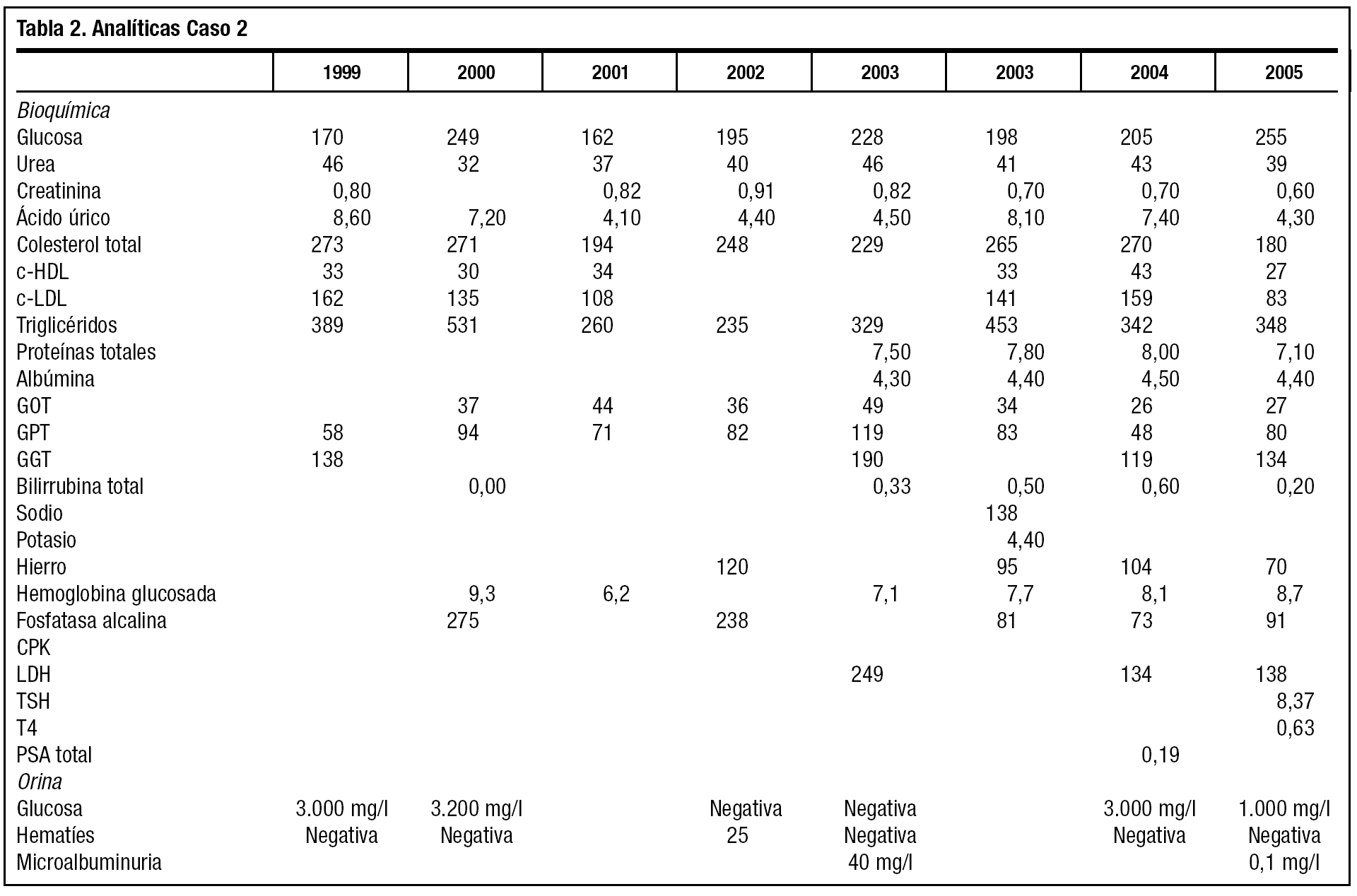
La tabla 2 muestra las cifras bioquímicas, más alteradas que las del caso 1, a pesar de su mejor adherencia al tratamiento farmacológico y la dieta. El paciente deja de fumar y restringe la ingesta enólica. La diabetes se trata de controlar asociando antidiabéticos. No tolera la metformina y la acarbosa y rechaza totalmente la insulina. Se pauta tratamiento con repaglinida y glimepirida, y tras comenzar tratamiento con estatinas se cambian a genfibrozil, por ser más acusadas la hipertrigliceridemia.
A finales de 2004 el hallux valgus es muy incapacitante y se envía a Traumatología realizando la extirpación y transposición tendinosa. El paciente presenta dehiscencia de sutura, y tras cicatrización por segunda intención evoluciona a úlcera vascular tórpida de unos 4 cm de diámetro que precisa cura diaria, retirada de esfácelos, vendaje y tratamiento antibiótico oral y analgésico durante más de 3 meses. Al final, aparece exudación serosanguinolenta, por la que es derivado al hospital para descartar osteomielitis. El Servicio de Cirugía Plástica realiza cura oclusiva con injerto de piel de la región gemelar, con buena evolución. Durante su estancia hospitalaria Endocrinología pauta tratamiento con insulina, además de añadir ácido acetilsalicílico (100 mg) al resto del tratamiento. Actualmente ambos casos han sido enviados a la Inspección médica para iniciar trámite de incapacidad, ya que todo el conjunto de sus patologías limitan severamente su capacidad laboral y autonomía.
DISCUSION
El SW es una muy rara patología hereditaria, de la que existen escasas referencias bibliográficas en nuestro país. Por ello sorprende el hallazgo de 2 casos en una población de apenas 400 habitantes.
La Atención Primaria brinda, desde el seguimiento longitudinal del paciente, un enfoque integrador de los diversos síntomas que van apareciendo. Con la presentación del caso tratamos de hacer pensar en esas enfermedades raras, algunas de sospecha fundamentalmente clínica, y cuyo seguimiento y control de los factores de riesgo sólo puede realizarse satisfactoriamente desde la Atención Primaria. Además, consideramos interesante para que nuestro trabajo sea más estimulante, tener en cuenta la existencia de estas enfermedades raras en nuestra actividad diaria.
Correspondencia:
A. Acevedo Gragera
C/ Madrid, 30. 06480 Montijo. Badajoz. España.
Correo electróncio: a.acevedog@terra.es
Recibido el 26-04-2005; aceptado para su publicación 25-11-2005.
