







INTRODUCCIÓN
En 1989 Darier y White describieron por primera vez la enfermedad.
Darier la denominó "psorospermosis folicular vegetante" y White "queratosis folicular". Es una genodermatosis poco frecuente caracterizada por un desorden de la queratinización. Se hereda de forma autosómica dominante con el gen defectuoso localizado en el brazo largo del cromosoma 12 (12q23-24), con una penetrancia muy alta superior al 95%1-5 y nuevas mutaciones. Afecta a ambos sexos por igual1,2.
Su severidad aumenta gradualmente con la edad. Empeora durante el verano debido al calor y la humedad y puede exacerbarse por exposición a la luz ultravioleta y por traumatismos mecánicos.
La etiología se desconoce pero el estudio microscópico sugiere un defecto de la síntesis, organización o maduración del complejo tonofilamento-desmosoma de las células de la capa basal que determina una queratinización anormal.
Cursa con la formación de pápulas queratósicas inicialmente de pequeño tamaño, ásperas, múltiples, de color rosado a bronceado. Con el tiempo las lesiones se recubren con una costra amarillenta y escamosa que al desprenderla deja una depresión. Al confluir, forman placas papilomatosas y verrucosas de aspecto grisáceo, como "sucio", que se distribuyen de forma simétrica4,5.
Se inicia generalmente entre los 6 y los 20 años de edad. Las lesiones predominan en las áreas seborreicas (cuello, pabellones auriculares, pliegue nasolabial, frente, cuero cabelludo, hombros, zona alta del tórax, espalda y zonas de flexión de las extremidades).
Existe afectación en áreas sin folículos:
Palmoplantar (queratosis puntiformes conocidas como acroqueratosis verruciforme de Hopfy).
Ungueal (muchas veces es la primera manifestación de la enfermedad, pero rara vez son las únicas afectadas). Las uñas son frágiles y estriadas (onicorrexis), con una muesca distal en forma de V. De forma excepcional se observa hiperqueratosis periungueal y perionixis inflamatoria.
Mucosas tanto orales (pápulas blancas con una depresión central localizadas en mejillas, paladar blando, duro y encías denominadas lesiones "en empedrado") como genitales, e incluso esofágicas6-8.
El grado de afectación es variable, desde lesiones banales imperceptibles hasta formas generalizadas. Existen formas atípicas de la enfermedad: ampollosas, hipertróficas, zoniformes o segmentarias (fenotipo I: existe una afectación localizada y la piel restante es completamente normal; fenotipo II: existe una afectación localizada en combinación con otra difusa).
Histológicamente encontramos una disqueratosis en la epidermis alta. En el estrato espinoso y granuloso hay unas células con núcleo picnótico con halo eosinófilo (cuerpos redondos). En el estrato córneo se encuentran células con núcleo alargado y escaso citoplasma (granos). También podemos observar en ciertas zonas una acantólisis suprabasal, acantosis, papilomatosis e hiperqueratosis.
No existe ningún tratamiento curativo. Para prevenir las crisis se pueden dar algunos consejos como evitar el sol, usar ropa de algodón, aplicar emolientes y evitar la transpiración.
En casos leves se recomienda el uso de queratolíticos tópicos y en casos severos retinoides orales9,10. Otras opciones de tratamiento son electrocirugía, láser, dermoabrasión y cirugía para las lesiones vegetantes grandes1-3.
CASO CLÍNICO
Un paciente de 30 años sin antecedentes médico-quirúrgicos de interés acude a la consulta por la presencia de múltiples pápulas de pequeño tamaño, ásperas al roce, de tonalidad marrón oscuro, localizadas principalmente en las superficies extensoras de los brazos y la zona superior de la espalda (figs. 1 y 2).
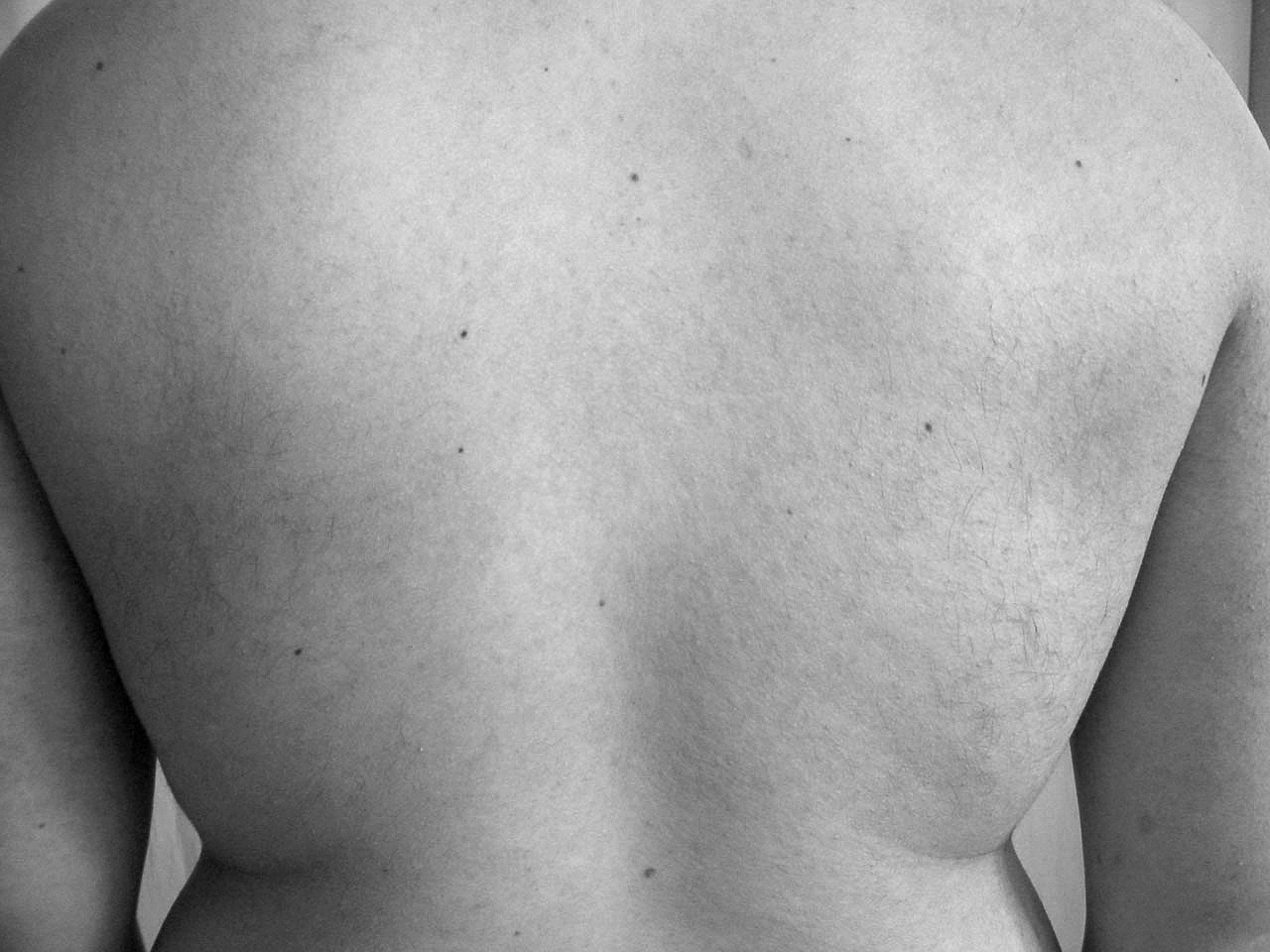
Figura 1. Presencia de lesiones pápulo-queratósicas al nivel de la espalda.
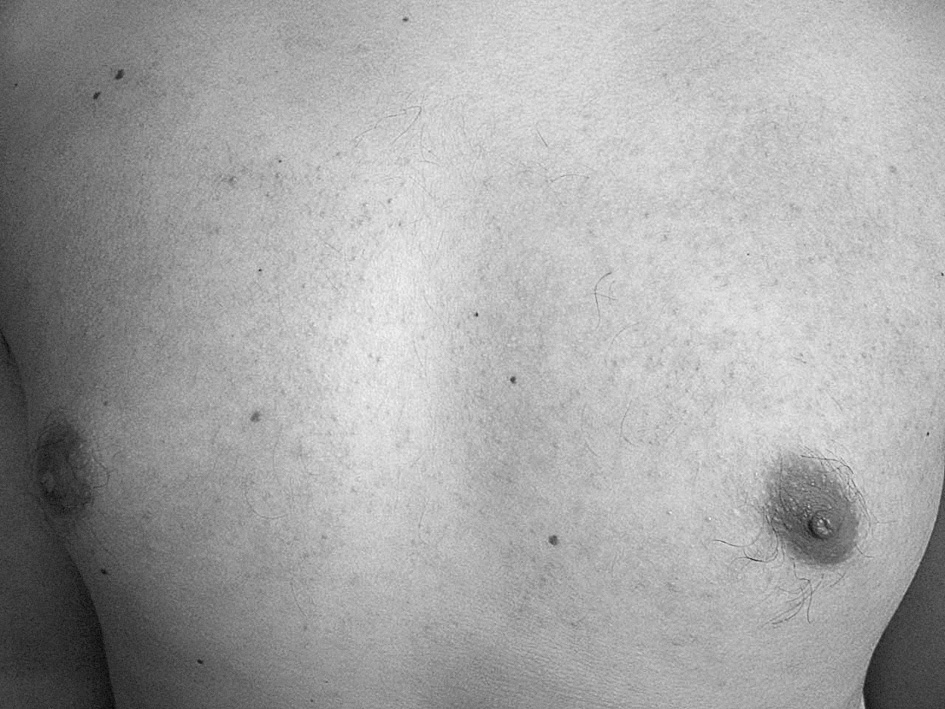
Figura 2. Presencia de pápulas queratósicas al nivel de la región superior del tórax.
El paciente refería tener estas lesiones desde hacía muchos años de evolución, pero notaba que iban empeorando progresivamente (sobre todo con el sol).
En la exploración física objetivamos la presencia de multitud de pápulas hiperqueratósicas e hiperpigmentadas de 0,5 mm, pruriginosas y malolientes en las localizaciones referidas (fig. 3).
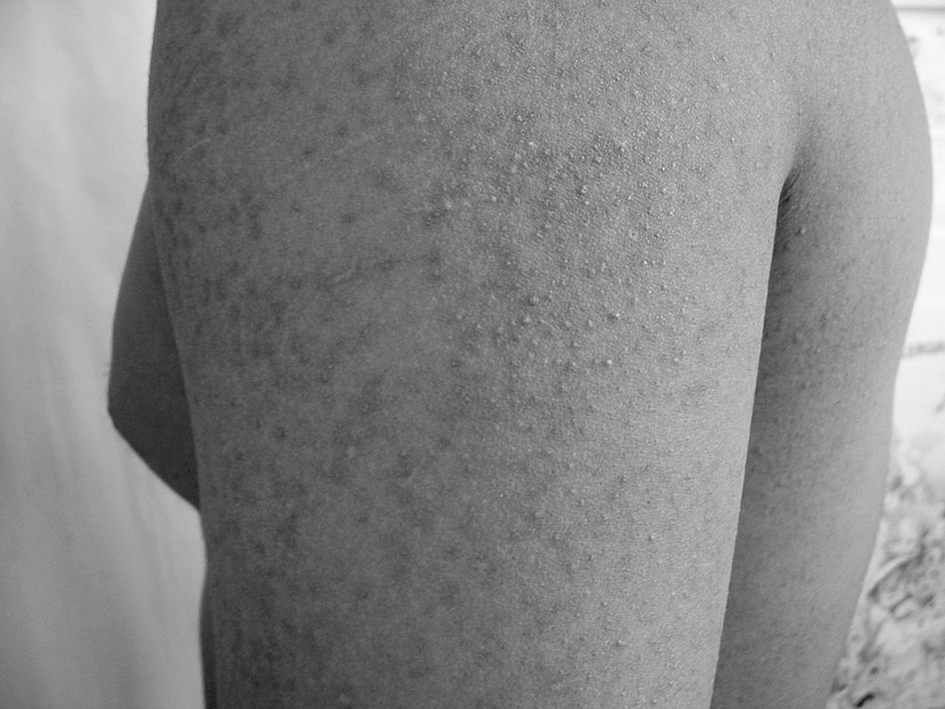
Figura 3. Presencia de lesiones pápulo-queratósicas de miembro superior izquierdo (imagen de lesiones más detallada).
No presentaba afectación palmo-plantar, ni ungueal, ni afectación de mucosas.
Al interrogatorio, como antecedentes familiares de interés, su padre presentaba las mismas lesiones, pero con un patrón mucho más acentuado.
Se le recomendó evitar el sol, así como la instauración de tratamiento con queratolíticos tópicos.
Se hace el diagnóstico presuntivo de enfermedad de Darier y se realiza estudio histopatológico de las lesiones. Los cortes muestran una epidermis con hiperqueratosis, tapones córneos y cuerpos redondos a nivel de la granulosa, así como presencia de células acantolíticas y granos. En la dermis superficial se observa papilomatosis e infiltrado linfohistocitario perivascular discreto.
El diagnóstico es compatible con la enfermedad de Darier.
DISCUSIÓN
El cuadro clínico se corresponde a otras series de casos publicados en la literatura revisada. La enfermedad de Darier-White o queratosis folicularis es una enfermedad pápulo escamosa que normalmente se manifiesta en la infancia (gradualmente progresiva en severidad en relación con la edad). Se hereda de forma autosómica dominante en ambos sexos por igual. En este caso se confirmó por recogerse en el árbol genealógico del enfermo al menos un sujeto en las últimas dos generaciones (su padre).
Se detectan múltiples pápulas hiperqueratósicas, rasposas, pardo-grisáceas o rojizas en relación con los folículos pilosos, pero existe afectación en áreas sin folículos como son la mucosa oral, las palmas y las plantas. De forma ocasional puede afectar a la conjuntiva y a la córnea pudiendo asociarse a cataratas2,3.
Tiene una prevalencia de 1/100.000. Se calcula una incidencia de 4 nuevos casos por millón de habitantes cada 10 años.
Resulta importante establecer el diagnóstico por la anamnesis (detectar los casos familiares) y el cuadro clínico (curso recurrente de las lesiones), así como lo concluyente del estudio histopatológico necesario para la confirmación, ya que las lesiones pápulo-costrosas pueden estar presentes en otras patologías.
En nuestro caso destacamos la demora en realizar el diagnóstico y las diferentes tentativas de tratamientos que, en su mayoría, exacerbaron el cuadro.
Las enfermedades asociadas son infrecuentes. Predominan los trastornos neurológicos (epilepsia, retraso mental, encefalopatía y atrofia cerebral) y manifestaciones psiquiátricas (trastornos del humor, depresión, esquizofrenia y psicosis maníaco depresiva).
Pueden aparecer complicaciones infecciosas con una especial sensibilidad a las infecciones virales, la más grave es la pustulosis varioliforme de Kapossi-Juliusberg.
El diagnóstico de confirmación se hace mediante biopsia, debido a que tanto en la enfermedad de Hailey-Hailey, como en la dermatosis acantolítica transitoria (enfermedad de Grover) y enfermedad de Galli-Galli, existe acantólisis suprabasal con proliferación ascendente de vellosidades en las ampollas. En la enfermedad de Darier se observan alteraciones típicas que consisten en hiperqueratosis, formación de hendiduras suprabasales, células epidérmicas disqueratósicas y cuerpos redondos6,7,10.
Se plantean diagnósticos diferenciales con la enfermedad de Hailey-Hailey (pénfigo crónico benigno familiar), enfermedad de Grover (dermatosis acantolítica transitoria o persistente) y enfermedad de Galli-Galli.
El pénfigo crónico benigno familiar es una dermatosis acantolítica hereditaria de transmisión autosómica dominante, y generalmente comienza en la adolescencia. Se caracteriza por la formación de lesiones eritematosas, erosivas con grietas y fisuración en la superficie localizadas fundamentalmente en la parte posterior del cuello, axilas, regiones submamarias e inguinales. Se produce con frecuencia una sobreinfección bacteriana o candidiásica. Puede establecer confusión con el intértrigo, la candidiasis y la dermatitis de contacto. Esta entidad es debida a una alteración genética en la regulación del calcio, necesaria para la correcta unión entre las proteínas de los desmosomas. Las erupciones empeoran en los meses de verano.
La enfermedad de Grover, o dermatosis acantolítica transitoria, es una dermatosis pruriginosa poco frecuente que afecta a varones de mediana edad, y consiste en la formación de grupos de lesiones papulosas, papulovesiculosas independientes, hiperqueratósicas, rojizas, escamo-costrosas y pruriginosas, con sensación de papel de lija a la palpación. Se localizan fundamentalmente en el tronco. Desde el punto de vista histopatológico encontramos una separación de las células íntimamente conectadas en las capas más exteriores de la piel (acantólisis). Su causa es desconocida, pero se cree relacionada con un traumatismo en la piel dañada por el sol. Los factores precipitantes suelen ser el exceso de calor, la fiebre o la exposición excesiva al sol. La enfermedad suele ser autolimitada.
La enfermedad de Galli-Galli es una genodermatosis rara, que se presenta en forma de placas reticulosas pigmentarias con elementos pápulo-queratósicos parduscos diseminados, localizados principalmente en los grandes pliegues9-11.
Correspondencia: M. D. Rua Guillermo
Pº Artilleros s/n
28032 Madrid
Correo electrónico: lolarg@hotmail.com
Recibido el 08-05-06; aceptado para su publicación el 15-11-06.
