









Las iniciativas sobre integridad científica requieren recomendaciones específicas para ensayos clínicos aleatorizados (ECA).
ObjetivoElaborar un documento consenso internacional con recomendaciones específicas que fomenten la integridad de los ECA.
MétodosEl documento se desarrolló conforme la metodología específica en varios pasos: composición y activación de un grupo multidisciplinar e internacional de expertos; registro prospectivo de protocolo; síntesis de evidencia de 55 revisiones sistemáticas, encuestas Delphi modificada con reunión final de expertos para el desarrollo del consenso.
ResultadosParticiparon 30 expertos en diversas áreas (ensayos clínicos, ética, metodología, estadística, revisiones sistemáticas, legislación, representantes de pacientes y de la industria, miembros de comités de financiación, autores, editores y revisores de revistas) que representaban a 15 países de los cinco continentes. La tasa de respuesta a la encuesta Delphi fue del 86,7% (26/30 expertos). A través de la metodología de consenso aplicada, se consolidaron 81 recomendaciones (49 proporcionadas por los expertos, 41 generadas por la revisión sistemática y nueve apoyadas por ambas) abarcando todo el ciclo de vida de los ECA: aspectos generales (n=6), diseño y la aprobación (n=11), realización y seguimiento (n=19), comunicación de protocolos y resultados (n=20), fase de pospublicación (n=12) y futuras líneas de investigación y desarrollo (n=13).
ConclusiónSe espera que la aplicación de este conjunto de recomendaciones mejore la integridad de los ECA.
Science integrity initiatives require specific recommendations for randomised clinical trials (RCT).
ObjectiveTo prepare a set of statements for RCT integrity through an international multi-stakeholder consensus.
MethodsThe consensus was developed via multi-country multidisciplinary stakeholder group composition and engagement; evidence synthesis of 55 systematic reviews concerning RCT integrity; anonymised two-round modified Delphi survey with consensus threshold based on the average percent of majority opinions; and, a final consensus development meeting.
ResultsThere were 30 stakeholders representing 15 countries from 5 continents including trialists, ethicists, methodologists, statisticians, consumer representative, industry representative, systematic reviewers, funding body panel members, regulatory experts, authors, journal editors, peer-reviewers and advisors for resolving integrity concerns. Delphi survey response rate was 86.7% (26/30 stakeholders). There were 111 statements (73 stakeholder-provided, 46 systematic review-generated, 8 supported by both) in the initial long list, with 8 additional statements provided during the consensus rounds. Through consensus the final set consolidated 81 statements (49 stakeholder-provided, 41 systematic review-generated, 9 supported by both). The entire RCT life cycle was covered by the set of statements including general aspects (n=6), design and approval (n=11), conduct and monitoring (n=19), reporting of protocols and findings (n=20), post-publication concerns (n=12), and future research and development (n=13).
ConclusionImplementation of this multi-stakeholder consensus statement is expected to enhance RCT integrity.
La integridad en la investigación es un término que no dispone de una definición única a la fecha pero la esencia de los múltiples conceptos y términos relacionados con la misma puede resumirse en una conducta responsable que, mediante el cumplimiento de las normas éticas1–5, garantice la transparencia en todas las etapas de la investigación, diseño, ejecución y comunicación de resultados3. Las iniciativas existentes hasta la fecha en relación con la integridad6–8 solo proporcionan recomendaciones generales sobre cómo promover conductas responsables.
En la investigación sobre la efectividad de las intervenciones sanitarias, los ensayos clínicos aleatorizados (ECA) y las revisiones sistemáticas que sintetizan sus resultados se sitúan en el nivel más alto de validez científica. Garantizar la integridad de los ECA es una prioridad9–11. Las encuestas sobre este aspecto publicadas muestran unas elevadas tasas de prácticas cuestionables11,12 que, unidas al creciente número de acusaciones y retracciones relativas a la fabricación de datos13, provocan una crisis de confianza de los profesionales y del público en general en la validez de la investigación clínica. Sin embargo, no todos los casos se deben a una mala conducta deliberada14. Una mezcla de errores no intencionados, metodología defectuosa, falta de concienciación sobre la ética de la investigación, malas habilidades de redacción, presión para publicar, entre otras1,10,15–17, comprometen la integridad de los ECA. Además, hasta donde sabemos, aparte del Consejo Internacional de armonización de los requisitos técnicos para el registro de medicamentos de uso humano (ICH)18, las iniciativas para preservar la integridad de la investigación6–8 no son específicas de los ECA. Esto dificulta que las instituciones académicas y sanitarias, así como los organismos de financiación de la investigación y las organizaciones editoriales se centren en los ECA para mejorar sus requerimientos y desarrollar una normativa específica relativa a la integridad de los mismos. Existe una necesidad urgente de cohesión multidimensional en este ámbito19.
Para abordar la necesidad de un conjunto actualizado y específico de recomendaciones sobre la integridad relativa a la conducta responsable en el desarrollo y publicación de ECA, desarrollamos un documento de consenso internacional, multidisciplinar, centrado en la transparencia necesaria en cada una de las fases de un ensayo como son la planificación, ejecución y difusión de resultados.
MétodosEl documento de consenso internacional sobre la integridad de los ECA se desarrolló siguiendo la metodología específica para este tipo de trabajos20–24, en varios pasos: a) composición y activación de un grupo multidisciplinar de expertos de varios países (a partir de agosto de 2021); b) síntesis de evidencia de revisiones sistemáticas relacionadas con la integridad de los ECA;19 c) registro prospectivo (https://osf.io/bhncy, 3 de diciembre de 2021), la encuesta Delphi modificada anónima en dos rondas (primera ronda: distribuida entre los participantes el 29 de enero y analizada el 6 de febrero de 2022; segunda ronda: distribuida entre los participantes el 8 de febrero y analizada el 18 de febrero de 2022); y, d) una reunión final de desarrollo de consenso (22 de febrero de 2022). El conjunto de datos brutos se puso a disposición del público en una plataforma de acceso libre (https://osf.io/92ahr) el 27 de junio de 2022.
Composición de un grupo multidisciplinar internacional de expertosEn agosto de 2021, seis meses antes de la reunión de consenso propuesta, se compuso cuidadosamente un grupo internacional de expertos. Se seleccionó a los miembros en función de sus conocimientos y experiencia para abarcar todos los aspectos críticos del ciclo de vida de los ECA. Nuestro enfoque utilizó la técnica de «bola de nieve», que detuvo la búsqueda de nuevos participantes una vez cubiertos todos los aspectos relevantes de los ECA25. Con esta técnica, a los expertos potenciales inicialmente contactados, se les pidió su aportación para identificar a otros miembros hasta cubrir todo el ciclo de vida de los ECA.
Un ensayo clínico se definió como un estudio en cuyo diseño se asigna aleatoriamente a participantes humanos a una o más intervenciones y donde existe un seguimiento de las variables de resultado para determinar el efecto de las intervenciones9.
Los expertos seleccionados incluían representantes de sociedades profesionales relevantes, de pacientes, público y consumidores, así como profesiones sanitarias relacionadas; investigadores, estadísticos y metodólogos; miembros y revisores de comités de ética, comités de monitorización y financiación; revisores y editores de revistas biomédicas. Se contactó con ellos directamente por correo electrónico (la lista de expertos y sus funciones se pueden consultar en la tabla 1). Nos aseguramos de que ninguno de los participantes tuviera publicaciones de ECA sometidas a una retracción o cuya validez estuviera siendo cuestionada mediante lo que en inglés se denomina como una «expression of concern» activa. Todos los expertos declararon explícitamente sus conflictos de intereses utilizando el formulario de declaración uniforme del Comité Internacional de Editores de Revistas Médicas (ICMJE) (Apéndice 1). Un miembro sin derecho a voto en las rondas Delphi (DM), sin experiencia en ECA, fue invitado al grupo para asesorar sobre la metodología de consenso y el lenguaje a utilizar.
Funciones y filiaciones de los participantes en el Documento de consenso internacional sobre la integridad de los ensayos clínicos
| Nombre | Papel de los autores | Filiación | ORCID ID |
|---|---|---|---|
| Yacoub Khalaf | Conceptualización, co-organizador, supervisión, contribución científica, revisión, edición y participante | Guy's & St Thomas’ Hospital Foundation Trust, Reino Unido | 0000-0002-5642-7367 |
| Khalid Saeed Khan | Conceptualización, co-organizador, supervisión, contribución científica, revisión, edición y participante | University of Granada; CIBERESP. España | 0000-0001-5084-7312 |
| Mohamed Fawzy | Conceptualización, metodología, administración del proyecto, contribución científica y participante | IbnSina, Banon Amshaj y Qena IVF Centres, Egipto | 0000-0001-8756-3612 |
| Patrick Chien | Contribución científica, validación, redacción, revisión y edición y participante | RUMC, Penang, Malasia | 0000-0002-5998-9592 |
| Aurora Bueno-Cavanillas | Contribución científica, metodología, validación y participante | Universidad de Granada; CIBERESP. España | 0000-0002-0649-3016 |
| Maria Núñez-Núñez | Redacción, curación de datos y participante | Hospital Universitario San Cecilio; Ibs Granada; CIBERESP. España | 0000-0002-2633-4207 |
| Marta Maes-Carballo | Redacción, curación de datos y participante | Complexo Hospitalario de Ourense; Hospital Público Verín. España. | 0000-0002-4852-5100 |
| Gamal Serour | Contribución científica, validación, representante de EFSS y participante | Al-Azhar University y Egiptoian IVF-ET Centre, Egipto | 0000-0002-0067-7850 |
| Mohamed Aboulghar | Contribución científica, validación, representante de MEFS y participante | Cairo University y Egiptoian IVF-ET Centre, Egipto | 0000-0002-3935-6501 |
| Gerben Ter Riet | Contribución científica, validación y participante | Amsterdam University, Paises Bajos | 0000-0002-2231-7637 |
| Javier Zamora | Contribución científica, análisis de datos, redacción y participante | Hospital Ramón y Cajal, IRYCIS. Madrid, España y Birmingham University, Reino Unido | 0000-0003-4901-588X |
| Jeffery Yrews | Contribución científica y participante | BD Integrated Diagnostic Solutions, Estados Unidos | 0000-0003-2416-0490 |
| Hassan Sallam | Contribución científica, y representante de ERC-RCOG y participante | Alexyria University, Egipto | 0000-0003-1308-6280 |
| Jack Wilkinson | Contribución científica y participante | Centre of Biostatistics, Manchester, Reino Unido | 0000-0003-3513-4677 |
| Hazem Abdelghaffar | Contribución científica y participante | Sohag University, Egipto | No disponible |
| Jacek Walczak | Contribución científica y participante | Centre of Excellence in Systematic Reviews, Central y Eastern Europe, CERTARA, Poly | 0000-0003-4965-0461 |
| Tayyiba Wasim | Contribución científica y participante | Services Institute of Medical Sciences, Services Hospital, Lahore, Pakistan | 0000-0003-2444-9817 |
| Ngawai Moss | Contribución científica y participante | University of London, Reino Unido | 0000-0001-9369-5072 |
| Hassan Maghraby | Contribución científica, representante de EFRE y participante | Alexyria University, Egipto | 0000-0003-3661-1594 |
| Jun Jim Zhang | Contribución científica y participante | Shangai Jiao Tong University School of Medicine, Shangai, China | No disponible |
| Ali Mahran | Contribución científica y participante | Assiut University, Egipto | 0000-0001-7870-4110 |
| Luciano Mignini | Contribución científica y participante | Hospital Escuela Eva Perón de Granadero Baigorria; Grupo Oroño. Argentina | 0000-0002-7783-9088 |
| Mahmoud Abdelaleem | Contribución científica y participante | Assiut University, Egipto | 0000-0003-3942-9325 |
| Mohamed Bedaiwy | Contribución científica y participante | University of British Columbia, Canada | 0000-0002-3454-8555 |
| Chris Hartgerink | Contribución científica y participante | Liberate Science GmbH, Alemania | 0000-0003-1050-6809 |
| Mohamed Sabry | Contribución científica y participante | Sohag University, Egipto | 0000-0002-8206-2074 |
| Mohamed Yahya AbdelRahman | Contribución científica y participante | Sohag University, Egipto | 0000-0002-0136-512x |
| Gian Carlo Di Renzo | Contribución científica y participante | University of Perugia, Perugia, Italia | 0000-0003-4467-240X |
| Zahida Qureshi | Contribución científica y participante | University of Nairobi, Kenia | 0000-0003-4223-3227 |
| Abdullah Alkhenizan Alkhenizan | Contribución científica y participante | Al Faisal University, Arabia Saudi | 0000-0002-0269-5200 |
| David Mortimer | Asesor, metodología consenso y apoyo a la redacción | University of Dundee, Escocia, Reino Unido y Oozoa Biomedical Inc, Canada | 0000-0002-0638-2893 |
CIBERESP: Epidemiology and Public Health Networking Biomedical Research Centre; EFRE: Egyptian Foundation of Reproductive Medicine and Embryology; EFSS: Egyptian Fertility and Sterility Society; ERC-RCOG: Egyptian Representative Committee of Royal College of Obstetricians and Gynaecologists; IRYCIS: Ramon y Cajal Institute for Biohealth Research; MEFS: Middle East Fertility Society.
Se seleccionó a dos miembros del grupo como coorganizadores (KSK e YK), encargados de supervisar el muestreo de «bola de nieve» y de garantizar que todos los expertos se identificaran con el alcance y el contenido del consenso, involucrándolos en discusiones, debates constructivos y resolución de desacuerdos. Tras aceptar la invitación, se celebraron entrevistas telemáticas o telefónicas con los expertos para informarles de los objetivos del proyecto y pedirles su aportación a las recomendaciones sobre integridad de los ECA.
Revisión sistemática paraguas de la literatura para la generación de recomendaciones basadas en evidenciaPara la creación de la lista inicial de recomendaciones, se llevó a cabo una revisión de las revisiones sistemáticas sobre la integridad de la investigación de los ECA. La revisión paraguas registrada prospectivamente (https://osf.io/3ursn) se llevó a cabo con una estrategia de búsqueda exhaustiva en las principales bases de datos electrónicas (PubMed, Scopus, Cochrane Library y Google Scholar) desde sus inicios hasta noviembre de 2021 para capturar la literatura gris y aquella revisada por pares. La estrategia de búsqueda y selección de artículos incluidos en la revisión, así como la extracción de datos, los métodos para evaluar la calidad metodológica y la síntesis de los resultados fueron publicados en paralelo a este consenso19.
A partir de los resultados de la revisión, un grupo seleccionado de cuatro expertos (AB, PC, MF y KSK) redactó recomendaciones claras, precisas y ejecutables. Dicho proceso de redacción se pilotó inicialmente con siete revisiones sistemáticas. Las deliberaciones de esta fase ayudaron a aclarar la distinción entre las conclusiones de las revisiones y las recomendaciones resultantes. Cada miembro de este grupo las redactó primero de forma independiente, con el objetivo de realizar una acción por afirmación, y luego se finalizaron mediante un debate.
Encuesta Delphi modificadaLas recomendaciones proporcionadas por los expertos se añadieron a las generadas a partir de la revisión de la literatura para crear la lista inicial para la encuesta de consenso Delphi modificada. Dicha encuesta fue enviada a los 30 expertos con derecho a voto que utilizaron una herramienta electrónica online (www.surveymonkey.com) para completarla. Para evaluar el nivel de acuerdo con el contenido de cada recomendación se empleó una escala de siete puntos comprendida entre «totalmente de acuerdo» y «totalmente en desacuerdo», con «de acuerdo», «algo de acuerdo», «ni de acuerdo ni en desacuerdo», «algo en desacuerdo» y «en desacuerdo» incluidas como las opciones de escala para las respuestas. Se utilizó la misma escala en las dos rondas de encuestas realizadas el 30 de enero y el 9 de febrero de 2022. La suma de las respuestas «totalmente de acuerdo» y «de acuerdo» se usó para calcular un índice de acuerdo para la inclusión de cada recomendación individual. Las respuestas de los expertos se mantuvieron anónimas durante todo el proceso.
Para determinar el umbral o punto de corte para la inclusión de las recomendaciones, utilizamos un método objetivo, el porcentaje medio de opiniones mayoritarias (APMO)24. Para este cómputo, se consideró que una recomendación estaba de acuerdo si la mayoría (> 50%) de los expertos respondía «totalmente de acuerdo» o «de acuerdo» en la escala de siete puntos. Una recomendación se consideró en desacuerdo si la mayoría (> 50%) de los participantes respondió «en desacuerdo» o «muy en desacuerdo» en la misma escala. El umbral de consenso de la APMO se calculó como: la suma de las recomendaciones mayoritariamente de acuerdo y mayoritariamente en desacuerdo/el número total de respuestas recibidas x 100%. Se consideró que las recomendaciones por encima del umbral APMO habían alcanzado el consenso para su inclusión. Para las recomendaciones individuales que lograron el consenso en cada ronda, calculamos la fuerza del acuerdo entre los participantes utilizando el rango intercuartílico (IQR)23.
El IQR fue la diferencia entre el primer y el tercer cuartil de las respuestas de los expertos en la escala de siete puntos y se interpretó de la siguiente manera: IQR 0 (> 50% de los expertos dieron las mismas respuestas) indicaba muy buena fuerza de acuerdo; IQR 1 (el rango de respuestas de> 50% de los expertos era ≤ 2 puntos de la escala) indicaba buena fuerza de acuerdo; IQR ≥ 2 (el rango de respuestas de> 50% de los expertos era> 2 puntos de la escala), indicaba pobre fuerza de acuerdo. Como análisis de sensibilidad, se empleó un umbral de aprobación arbitrario del 70%. Los resultados se analizaron usando el software Stata v16 los días 6 y 18 de febrero de 2022 (StataCorp. 2019, College Station, TX: StataCorp LLC, EE. UU.).
Las recomendaciones que no alcanzaron el consenso en la primera ronda utilizando el umbral APMO se fusionaron con otras nuevas proporcionadas por los expertos y se sometieron a la segunda ronda de la encuesta Delphi modificada. Se mejoró la redacción de las afirmaciones que no alcanzaron el consenso por falta de claridad en el lenguaje. Las recomendaciones que contenían información similar se fusionaron para evitar duplicados. La tasa de acuerdo de la primera ronda se proporcionó en la segunda ronda de la encuesta junto con las referencias a las revisiones sistemáticas incluidas que apoyaban las recomendaciones generadas mediante la síntesis de la evidencia. La edición menor, la fusión de recomendaciones y el enfoque estadístico de la segunda ronda fueron los mismos que los utilizados en la primera. Las recomendaciones que no alcanzaron el consenso se sometieron a votación en la reunión final de desarrollo del consenso.
Para consolidar el conjunto de recomendaciones provisionales, un grupo de expertos (AB, KSK, MNN, PC y MF) evaluó aquellas que habían alcanzado el consenso en cuanto a duplicidades exactas o inexactas y claridad de significado. En los casos en que la duplicidad era prácticamente exacta, se creó una recomendación única, introduciendo solo pequeños cambios de redacción para aclarar o mejorar el significado deseado. No se introdujeron modificaciones importantes de redacción en ninguna de las recomendaciones que habían alcanzado el umbral de consenso. Aquellas sin consenso se revisaron del mismo modo con vistas a mejorar la claridad de su significado y a ayudar en la votación posterior.
Así pues, las recomendaciones originales pueden haber sido objeto de pequeños cambios de redacción o de fusión con otras, varias veces a lo largo de las distintas rondas de consenso para mejorar su claridad. La lista de recomendaciones resultantes del proceso anterior, tanto las que habían alcanzado el consenso como las que no, se tabuló y se distribuyó a todos los participantes con las calificaciones de acuerdo y las referencias de apoyo a las revisiones para la reunión de desarrollo del consenso.
Reunión de desarrollo del consensoTodos los expertos fueron invitados a la reunión y asistieron durante todo el día 24 participantes en persona, seis de forma virtual y DM en persona como asesor. El conjunto provisional de recomendaciones tabuladas anteriormente se compartió con los expertos junto con un borrador inicial de este manuscrito. En la reunión, celebrada en El Cairo (Egipto) el 22 de febrero de 2022, se debatieron individualmente las recomendaciones clasificadas como no consensuadas en la encuesta Delphi de dos rondas. Los expertos decidieron la tasa de acuerdo que se utilizaría como umbral de exclusión y votaron de forma anónima mediante un sistema electrónico (https://zoom.us/) para seleccionar las recomendaciones del conjunto final.
Se acordó con el grupo de expertos el desglose de las recomendaciones cubriendo las distintas etapas del ciclo de vida de los ECA. Se incluyeron los siguientes apartados: aspectos generales, diseño y aprobación, realización y seguimiento, comunicación de protocolos y resultados, fase de pospublicación, futuras líneas de investigación y desarrollo. Con el conjunto final de recomendaciones, se proporcionó la calidad de la evidencia evaluada mediante una A MeaSurement Tool to Assess systematic Reviews- 2 (AMSTAR-2) modificada26 para las que estuvieron respaldadas por revisiones sistemáticas.
Participación de pacientes y ciudadanosUn experto que acudió como representante de los pacientes (NM) fue incluido en el grupo de consenso para aportar información como participante en el ensayo. Tres expertos (NM, ABC y KSK) tenían experiencia previa en la participación en ECA de pacientes, público y consumidores27,28 (fig. 1). Además, tres revisiones sistemáticas incluidas en la síntesis de la evidencia abordaron cuestiones de integridad de los ECA relacionadas con la participación de pacientes, público y consumidores29–31. Este manuscrito se ha elaborado de acuerdo con las directrices Guidance for Reporting Involvement of Patients and the Public-2 (GRIPP-2) (Apéndice 2)32.
Resultados
Participaron 30 expertos (tabla 1) con derecho a voto procedentes de 15 países de cinco continentes, entre los que se incluían especialistas en ensayos clínicos, ética, metodólogos, estadísticos, representantes de pacientes y de la industria, expertos en revisiones sistemáticas, miembros de comités de financiación, expertos en normativa, autores, editores de revistas, revisores y asesores de conflictos relacionados con la integridad de la investigación. El grupo de expertos reunía una amplia y adecuada experiencia, basada en la autoevaluación, que abarcaba de manera exhaustiva todos los aspectos del ciclo de vida de la investigación de ECA, desde el desarrollo del protocolo hasta la transferencia de conocimientos (fig. 1). En su selección se tuvo en cuenta toda la experiencia profesional pertinente de los especialistas, no solo la filiación en el momento del desarrollo de este consenso, y se incluyó finalmente una cobertura geográfica de 22 países y seis continentes (fig. 2).
 según el lugar de filiación en el momento del consenso; y (B) según la experiencia profesional relevante (solo se han comunicado los datos de los miembros con derecho a voto).")
Distribución geográfica del grupo de expertos del documento de consenso internacional sobre la integridad de los ensayos clínicos: (A) según el lugar de filiación en el momento del consenso; y (B) según la experiencia profesional relevante (solo se han comunicado los datos de los miembros con derecho a voto).
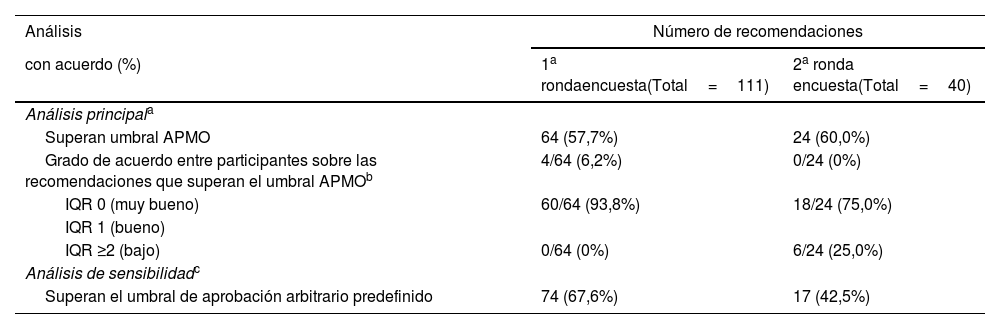
La lista inicial de 111 recomendaciones (73 aportadas por los expertos, 46 generadas mediante síntesis de la evidencia19 y ocho respaldadas por ambas) se sometió a consenso mediante la encuesta Delphi modificada (fig. 3). En la primera ronda de la encuesta respondieron 26 de 30 (86,7%) encuestados, y 64 recomendaciones fueron valoradas por encima del umbral del 76,5% de la APMO para el consenso. La fuerza del acuerdo entre los expertos fue buena o muy buena en todas ellas (tabla 2). Las 47 recomendaciones restantes, junto a siete nuevas aportadas por los participantes, se sometieron a revisión. Tras fusionar los duplicados exactos e inexactos, se sometieron 40 recomendaciones a la segunda ronda de encuestas, en la que respondieron 26 de 30 (86,7%) encuestados y 24 recomendaciones fueron calificadas por encima del umbral de consenso de la APMO del 68,4%. La fuerza del acuerdo entre los expertos fue buena en 18 (75%) de ellas (tabla 2). Las 64 recomendaciones acordadas en la primera ronda de la encuesta Delphi modificada se fusionaron, eliminando duplicados exactos e inexactos, quedando 54, que junto a las 24 acordadas en la segunda ronda se llevaron a la reunión de desarrollo del consenso, así como las 16 recomendaciones para las que no se alcanzó un consenso tras la segunda ronda. El análisis de sensibilidad para el umbral de consenso utilizando el límite arbitrario predefinido del 70% mostró que el umbral de la APMO era más conservador en la primera ronda, lo que permitió reexaminar más recomendaciones (tabla 2).

Recomendaciones según los diferentes umbrales de acuerdo en el consenso internacional sobre la integridad de los ensayos clínicos
| Análisis | Número de recomendaciones | |
|---|---|---|
| con acuerdo (%) | 1a rondaencuesta(Total=111) | 2a ronda encuesta(Total=40) |
| Análisis principala | ||
| Superan umbral APMO | 64 (57,7%) | 24 (60,0%) |
| Grado de acuerdo entre participantes sobre las recomendaciones que superan el umbral APMOb | 4/64 (6,2%) | 0/24 (0%) |
| IQR 0 (muy bueno) | 60/64 (93,8%) | 18/24 (75,0%) |
| IQR 1 (bueno) | ||
| IQR ≥2 (bajo) | 0/64 (0%) | 6/24 (25,0%) |
| Análisis de sensibilidadc | ||
| Superan el umbral de aprobación arbitrario predefinido | 74 (67,6%) | 17 (42,5%) |
APMO: porcentaje medio de opiniones mayoritarias; IQR: rango intercuartílico.
En este cálculo, una recomendación se consideró con acuerdo, y por tanto se incluiría si la mayoría (> 50%) de los participantes respondió «totalmente de acuerdo» o «de acuerdo» en la escala de siete puntos. Por contrario, una recomendación se consideró en desacuerdo si la mayoría (> 50%) de los participantes respondió «en desacuerdo» o «muy en desacuerdo» en la escala de siete puntos. El umbral de aprobación de la APMO se calculó como: suma de recomendaciones mayoritariamente de acuerdo y mayoritariamente en desacuerdo/número total de respuestas recibidas×100%. Los umbrales de aprobación APMO fueron del 76,4% en la primera ronda Delphi y del 68,4% en la segunda ronda Delphi.
Rango intercuartílico (IQR) de las respuestas en la escala de siete puntos. En este cómputo, IQR 0 (> 50% participantes dieron las mismas respuestas) indicó muy buen grado de acuerdo; IQR 1 (> 50% participantes rango de respuestas fue ≤ 2 puntos de la escala) indicó buen grado de acuerdo; IQR ≥ 2 (> 50% participantes dieron respuestas> 2 puntos de la escala) indicó bajo grado de acuerdo.
Se incluyó una nueva recomendación proporcionada por los expertos, lo que elevó a 95 el total presentado en esta fase final. Al principio del procedimiento, el grupo de expertos aceptó que se excluirían las recomendaciones por debajo del umbral de acuerdo del 50%. Tras el debate, la fusión y la votación en la reunión de desarrollo del consenso, la lista final de preseleccionados incluía 81 recomendaciones (49 aportadas por los participantes, 41 generadas por la revisión sistemática; nueve incluidas en cada grupo por un doble respaldo). Del total, 32 (39,5%) eran únicamente basadas en la evidencia.
De las 41 recomendaciones basadas en la síntesis de la evidencia19, dos se fundamentaron en al menos una revisión sistemática de calidad alta-moderada29,33. Como se muestra en la tabla 3, se cubrió todo el ciclo de vida del ECA con recomendaciones relativas a aspectos generales (n=6), diseño y aprobación (n=11), realización y seguimiento (n=19), comunicación de protocolos y resultados (n=20), fase de pospublicación (n=12), futuras líneas de investigación y desarrollo (n=13).
Recomendaciones del documento de consenso internacional sobre la integridad de los ensayos clínicos (n=81)
| Recomendaciones finales consensuadas | Acuerdo (%)a | Fuente de informaciónb | ||
|---|---|---|---|---|
| Delphi 1a ronda(umbral 76,5%) | Delphi 2a ronda(umbral 68,4%) | Reunión de consenso | ||
| Aspectos generales | ||||
| 1. Las directrices y políticas sobre integridad de los ensayos clínicos deben ser explícitas y visibles, e ir acompañadas de un plan de implementación prospectivo. | 82,7c | RP | ||
| 2. Los investigadores de los ensayos, miembros de los comités de ética, editores y revisores de revistas deben recibir formación adecuada en metodología e integridad de la investigación. | 80,8c | RP,1-7 | ||
| 3. Los comités de ética deben estar acreditados y tanto los criterios de evaluación ética como los procedimientos de revisión deben estar armonizados a nivel regional, nacional e internacional. | 92,3c | 8,9 | ||
| 4. La documentación actualizada del estudio debe estar disponible durante todo el ciclo de vida de los ensayos. | 61,5 | 61,5 | 80,0 | RP |
| 5. Las revistas deben apoyar la adopción de prácticas de investigación responsables en el diseño, la realización, el análisis, la notificación y el archivo de los ensayos. | 88,5 | RP | ||
| 6. Las instituciones deben evitar una presión excesiva por publicar. | 76,9 | RP | ||
| Diseño y la aprobación | ||||
| 7. Debe obtenerse la aprobación del comité de ética en todos los ensayos, incluidos los que utilicen datos no-identificables. | 67,3c | 65,5c | 100 | 10,11,20,21 |
| 8. El consentimiento informado debe elaborarse con la participación de pacientes (o sus representantes) y público general. | 80,8 | 12,13,14,15,16 | ||
| 9. El consentimiento informado debe ser examinado y aprobado por el comité de ética. | 96,2 | 1,12,14 | ||
| 10. El consentimiento informado debe incluir explícitamente cómo se compartirán los datos no-identificables en el momento de la publicación y cómo se utilizarán para futuros análisis. | 73,1 | 65,4 | 96,4 | 17 |
| 11. Los ensayos clínicos deben priorizarse y dotarse de recursos en función de las necesidades locales de salud, estratégicas y culturales, especialmente en ensayos multinacionales que incluyan entornos de bajos recursos. | 69,2 | 69,2d | 1,12,18 | |
| 12. Los ensayos deben aprobarse de acuerdo con el marco ético y normativo local, especialmente en ensayos multinacionales que incluyan entornos de bajos recursos. | 76,9 | 1,12,18 | ||
| 13. En los ensayos multinacionales, las traducciones de los resultados comunicados por los pacientes deben tener en cuenta los aspectos culturales, incluidos los entornos de bajos recursos. | 84,6 | 19 | ||
| 14. La igualdad, la diversidad y la inclusión deben considerarse en el diseño de los ensayos para maximizar la extrapolación de los resultados. | 76,9 | RP | ||
| 15. La información sobre la estimación del tamaño de la muestra debe ser lo suficientemente detallada como para permitir su reproducción. | 92,3 | 24 | ||
| 16. Las variables resultado primarias y secundarias deben ser las internacionalmente aceptadas como «core outcomes» (desenlaces fundamentales) siempre que estén disponibles. | 80,8 | RP | ||
| 17. El protocolo del ensayo, incluida la aprobación ética, debe registrarse prospectivamente en un registro de ensayos de acceso abierto antes del reclutamiento de los participantes. Esta política debe incluirse en los reglamentos de las instituciones de investigación, los patrocinadores, las agencias de financiación y en las condiciones de los contratos de trabajo de los investigadores. | 78,9c,d,e | RP, 30,32,35 | ||
| Realización y seguimiento | ||||
| 18. El centro en el que se realiza el ensayo debe establecer medidas de evaluación que permitan mitigar los incumplimientos de la integridad, con el apoyo de los servicios locales de gobernanza de la investigación. | 88,5c | RP | ||
| 19. Durante la ejecución de un ensayo, debe promoverse la admisión de errores honestos o involuntarios sin temor a ser culpado. Una parte de esta política debe ser la formación. | 94,2c | RP | ||
| 20. Las estrategias innovadoras de reclutamiento deben estar impulsadas por los participantes y deben cumplir los principios éticos. | 88,5 | 15,25,26f | ||
| 21. Los datos recogidos de forma rutinaria deben validarse antes de su análisis y notificación. | 69,2 | 84,6 | RP, 20,27 | |
| 22. La supervisión del consentimiento informado debe formar parte de la auditoría del ensayo. | 92,3 | 10,13 | ||
| 23. Los miembros de los comités directivos de los ensayos y de los de monitorización de datos deben declarar cualquier posible conflicto de intereses. | 100 | RP | ||
| 24. Entre los miembros de los comités directivos de los ensayos deben figurar representantes de pacientes y público. | 69,2 | 65,4 | 79,3 | RP |
| 25. Las actas de las reuniones de los comités directivos del ensayo y de las de los de monitorización de datos deben estar disponibles cuando se soliciten. | 69,2 | 61,5 | 83,0 | RP |
| 26. Los estatutos del comité de monitorización de datos deben incluir la responsabilidad de la integridad de los datos. | 92,3 | RP,28 | ||
| 27. Para garantizar la integridad de los datos, debe implantarse una supervisión centralizada y una verificación selectiva de los datos de origen. | 80,8 | 29 | ||
| 28. Debe haber transparencia en el método o métodos de obtención de los datos faltantes en todas las fases del seguimiento y la elaboración de informes. | 96,2 | RP | ||
| 29. La finalización anticipada de un ensayo debe realizarse con la participación independiente de los comités directivo y de monitorización de datos. | 96,0 | RP | ||
| 30. Cualquier modificación del protocolo del estudio debe notificarse al registro del ensayo (incluyendo fechas). Los cambios importantes también requieren aprobación por el comité de ética. | 100 | RP | ||
| 31. El plan de análisis estadístico debe desarrollarse y publicarse al inicio o durante las primeras fases del ensayo, antes de que los investigadores dispongan de los datos. | 88,5 | RP | ||
| 32. Todos los análisis deben ser preespecificados desde el principio (el análisis de la variable resultado primaria y las variables secundarias, los análisis de subgrupos y los análisis de sensibilidad). | 84,6 | RP | ||
| 33. Debe haber un único resultado primario preespecificado; cuando hay múltiples resultados clave, deben considerarse estrategias de testeo válidas para mantener el error de tipo 1 dentro del límite aceptable del 5%. | 65,4 | 69,2d | RP | |
| 34. Los patrocinadores de ensayos clínicos deben estipular en los contratos con los investigadores la obligación de analizar y comunicar los resultados de acuerdo con el protocolo previamente registrado. | 42,3 | 57,7 | 88,0 | RP |
| 35. Las bases de datos para los ensayos clínicos deben incluir registros de acceso auditables y sistemas de gestión de permisos que impidan su edición o el acceso ilícito a los datos. | n/ag | n/ag | 100 | RP |
| 36. La integridad de los ensayos, como la calidad de la síntesis de evidencia, requiere evitar o minimizar los sesgos en la realización de los ensayos. | n/ag | 84,6 | RP | |
| Comunicación de protocolos y resultados | ||||
| 37. Se recomienda encarecidamente a los autores de ensayos que no presenten resultados a revistas depredadoras, y eviten las revistas poco transparentes o con integridad cuestionable. | 69,2 | 65,4c | 83,3 | 30 |
| 38. Las instrucciones de las revistas para los autores deben incluir de forma explícita y exhaustiva los requisitos de apertura y transparencia. | 84,6c | RP, 31,32,33,34 | ||
| 39. El sistema de presentación electrónica de las revistas debe facilitar el cumplimiento de las instrucciones para los autores relacionadas con la integridad. | 73,1 | 92,3 | RP | |
| 40. La redacción médica profesional podría facilitar el desarrollo de publicaciones más claras y concisas, que cumplan adecuadamente los requisitos de integridad. La utilización de este recurso debe quedar explicita en la publicación. | 61,5 | 69,2d | 36 | |
| 41. La rapidez de la revisión por pares y de la toma de decisiones editoriales debe estar equilibrada con la posibilidad de futuras quejas y retractaciones. | 65,4 | 65,4 | 83,3 | 37 |
| 42. La notificación de la aprobación del comité de ética y los detalles del consentimiento informado deben ser parte obligatoria de las directrices para describir un estudio y de las instrucciones para los autores. | 84,6c | 10,13, 14,17,38 | ||
| 43. El comité de ética que evaluó el ensayo o un comité independiente de monitorización de datos deben confirmar que el ensayo se ha realizado según lo previsto. | 61,6c | 69,5c | RP | |
| 44. La contribución de la autoría debe especificarse en el manuscrito de acuerdo con las directrices internacionales (taxonomía de roles de autoría). | 94,3c | RP,22,23 | ||
| 45. El protocolo del ensayo y el plan de análisis estadístico deben presentarse sin editar, junto con el conjunto de datos, la sintaxis estadística y los resultados analíticos. | 69,2 | 88,5 | RP,7,33 | |
| 46. La notificación de conflictos de intereses, fuentes de financiación y pagos recibidos por todos los autores debe hacerse de forma estandarizada. | 78,9c | RP,23,34,39,40,41 | ||
| 47. La declaración de conflictos de intereses, fuentes de financiación y pagos recibidos debe ser obligatoria para los revisores y editores de revistas científicas. | 88,5 | RP | ||
| 48. La información sobre la participación de pacientes y público en el ensayo debe ser obligatoria. | 76,9 | RP | ||
| 49. Los manuscritos deben prepararse de acuerdo con las directrices para la redacción de informes (p. ej., SPIRIT, CONSORT, GRIPP-2, etc.) y sus extensiones específicas para tipos de ensayos concretos (p. ej., ensayos de provocación en humanos, ensayos de intervenciones sociales y psicológicas, etc.). | 76,9c,d,h | RP,42,43, 47 | ||
| 50. Se deben realizar controles de plagio en el texto principal del artículo de forma sistemática. | 84,6 | 44 | ||
| 51. Los errores, las desviaciones del protocolo, las pérdidas durante el seguimiento, la información sobre datos faltantes y las soluciones aplicadas deben comunicarse de forma transparente. | 92,3 | 45,46,54 | ||
| 52. Debe ser obligatorio informar sobre los comités de monitorización de datos, su composición y sus responsabilidades. | 73,1 | 96,2 | 28 | |
| 53. Entre los ensayos realizados en varios idiomas, el uso de traducciones en los resultados reportados por los pacientes debe ser explícito. | 53,8 | 53,8 | 91,6 | 19 |
| 54. Los desenlaces primarios y secundarios deben vincularse obligatoriamente a los registrados prospectivamente. | 76,9 | 35 | ||
| 55. Debe eliminarse la posibilidad de tergiversar, exagerar o distorsionar por escrito los métodos, hallazgos, resultados y conclusiones. | 82,7c | RP | ||
| 56. Los puntos fuertes y las limitaciones de las cuestiones relacionadas con la integridad, así como cualquier desviación en el desarrollo de la metodología respecto a lo ideal, aunque haya sido inevitable, deben incluirse en el apartado de discusión del manuscrito. | 73,1 | 96,2 | RP | |
| Fase de pospublicación | ||||
| 57. La detección de infracciones de la integridad tras la publicación implica que el proceso científico ha fallado. La atención debe centrarse en la identificación de las causas y su corrección, facilitando un aprendizaje abierto y colectivo. | 76,9 | RP | ||
| 58. Las revistas tienen la responsabilidad de evaluar y conducir la revisión por pares previa a la publicación de manera que se minimice el riesgo de acusaciones de deshonestidad posteriores a la publicación. | 92,3 | RP | ||
| 59. Toda orientación relativa a los problemas de integridad posteriores a la publicación (p. ej., COPE https://publicationethics.org, https://doi.org/10.24318/o1VgCAih, https://doi.org/10.24318/cope.2019.2.4) debe insistir explícitamente en la responsabilidad de los investigadores de evaluar la integridad de la denuncia y apoyar a los autores de los ensayos. | 73,1 | 88,5 | RP | |
| 60. Las instituciones y las revistas deberían apoyar por igual al denunciante o denunciantes y al autor o autores en la tramitación de dichas denuncias, asumiendo la responsabilidad de proteger a los autores honestos contra el acoso. | 84,6c | RP | ||
| 61. Los autores de los ensayos deben responder a cualquier solicitud de explicación de una aparente discrepancia en los datos, si así lo requiere la revista, tanto durante las fases de revisión por pares como tras la publicación. Igualmente deben responder a las solicitudes de aclaraciones de los revisores sistemáticos durante la síntesis de la evidencia. | 92,3 | RP | ||
| 62. Los autores de los ensayos tienen la responsabilidad de conservar registros detallados de su estudio, incluido el protocolo original (con cualquier modificación posterior), la aprobación del comité de ética, los detalles del registro del ensayo, el conjunto de datos brutos anonimizados, la secuencia de randomización empleada, el plan estadístico, la sintaxis y los resultados de todos los análisis estadísticos y facilitarlos en caso de que se requieran para aclarar cualquier reclamación posterior a la publicación. | 80,8 | RP | ||
| 63. La declaración de conflictos de interés, fuentes de financiación y pagos recibidos debe ser obligatoria también para los denunciantes. | 84,6 | RP | ||
| 64. Las revistas deben actuar de forma imparcial y transparente en la gestión de los conflictos de interés de sus propios editores y asesores ante reclamaciones. | 80,8c | RP | ||
| 65. Debe permitirse que los autores de los ensayos, con la participación de su institución, proporcionen informes periciales independientes a la revista que investiga una reclamación. | 76,9 | RP | ||
| 66. Cuando se detectan errores honestos después de la publicación, debe publicarse una fe de errata. | 96,2 | RP | ||
| 67. Los comunicados de retractación deben ser claros e interpretables. | 88,5 | 48 | ||
| 68. La gestión posterior a la retractación de los ensayos con mala conducta demostrada debe basarse en un sistema que evite la citación continua y el uso indebido de los datos. | 96,2 | 48 | ||
| Futuras líneas de investigación y desarrollo | ||||
| 69. Debe evaluarse la eficacia educativa de la formación en materia de integridad. | 69,2 | 84,6 | 53f | |
| 70. Deben evaluarse los factores que influyen en la disposición de los participantes a dar su consentimiento para compartir datos. | 61,5 | 76,9 | 51,52 | |
| 71. Deben establecerse los requisitos mínimos para obtener un consentimiento informado adecuado. | 61,5 | 69,2 | 49 | |
| 72. Deben definirse los criterios y el nivel de auditoría de datos requerido durante la realización del ensayo. | 61,5 | 65,4 | 100 | 10,49 |
| 73. Deben aclararse las funciones de los comités de control de la integridad de los datos. | 69,2 | 80,8 | 28 | |
| 74. Debe determinarse el método o métodos más adecuados para establecer la autoría (contribución de los autores). | 65,4 | 88,5 | 50 | |
| 75. Deben desarrollarse modelos eficaces de revisión por pares para la evaluación de los ensayos. | 84,6 | 55 | ||
| 76. Deben desarrollarse controles automatizados del cumplimiento de las directrices de redacción de informes (p. ej., CONSORT, SPIRIT, GRIPP-2). | 80,8 | RP | ||
| 77. Para compartir los datos brutos, las revistas deben aclarar los requisitos, por ejemplo, secuencia de randomización, conjunto de datos originales anonimizados, códigos estadísticos, etc. | 69,3c | 92,3 | RP | |
| 78. Debe evaluarse la validez de los test de integridad aplicados tras el envío y/o publicación de los ensayos. | 65,4 | 84,6 | 44 | |
| 79. Debe desarrollarse una terminología de investigación común que permita evitar la publicación de información selectiva. | 57,7 | 53,8 | 86,9 | 54 |
| 80. Los procedimientos para la síntesis de la evidencia aportada por ensayos con datos publicados a nivel de estudio (no brutos) deben desarrollar métodos (p. ej., metaanálisis de subgrupos o meta-regresión) para evaluar las cuestiones de integridad de los ensayos. | n/ag | 69,2d | RP | |
| 81. Los procedimientos para la síntesis de la evidencia deben desarrollar métodos para acceder a los datos (brutos) a nivel de paciente a fin de maximizar la transparencia. | n/ag | 76,9 | RP | |
Nota: Para más detalles, véase la figura 3 y el archivo de intercambio de datos (https://osf.io/92ahr).
El acuerdo (%) para las rondas Delphi es el porcentaje de la suma de las respuestas «totalmente de acuerdo» y «de acuerdo» proporcionadas en la escala de siete puntos para la inclusión de cada recomendación individual por parte de los participantes. El acuerdo (%) durante la reunión de consenso es el porcentaje de votos emitidos a favor sobre el total de votos.
El porcentaje de acuerdo (78,9%, la mediana de 88,5%, 84,6%, 73,08% y 61,54%) representa los datos de una recomendación fusionada que contiene cuatro recomendaciones, dos aprobadas en la primera ronda (relacionadas con el registro prospectivo, 88,5% y 84,6%) y las otras dos aprobadas en la segunda ronda (relacionadas con la política, 73,08% y 61,54% en la primera ronda y superaron el umbral de aprobación en la segunda ronda con 80,77% y 69,23%). El grado de acuerdo entre los participantes para las recomendaciones incluidas en la segunda vuelta fue bajo en la primera vuelta y bueno/bajo en la segunda vuelta (véanse los detalles en Métodos y tabla 2).
n/a: no aplicable, la recomendación fue proporcionada por un participante después de la primera o la segunda ronda Delphi.
El porcentaje de acuerdo (76,9%, la mediana de 84,6% y 69,2%) representa los datos de una recomendación fusionada que contiene dos recomendaciones, una incluida en la primera ronda (relacionada con las guías de notificación, 84,6%) y la otra aprobada en la segunda ronda (relacionada con las extensiones específicas, 69,2% en la primera ronda y superó el umbral de recomendación en la segunda ronda con un 69,2%). El grado de acuerdo entre los participantes sobre la recomendación relativa a las prórrogas específicas fue bueno en la primera vuelta y pobre en la segunda (véanse los Métodos y la tabla 2 para más detalles).
Este consenso internacional de expertos proporciona la primera declaración de integridad específica para promover y proteger la integridad de los ECA. Se elaboró de forma sólida y exhaustiva, abarcando todo el ciclo de vida de los mismos. Las recomendaciones generales sobre la integridad de los ECA recalcan la necesidad de armonización y acción conjunta a escala mundial, aquellas relativas al diseño, la aprobación, la realización y el seguimiento de los ECA dejan claro que la integridad debe incluirse en todo el ciclo de vida de la investigación. Mientras que las responsabilidades de la comunidad editorial se abordan en las recomendaciones concernientes a la presentación de manuscritos, revisión por pares, presentación de informes y reclamaciones. Otras recomendaciones subrayan la necesidad de continuar la investigación y el desarrollo para avanzar en la conducta responsable de la investigación en los ECA. Redactado en un lenguaje sencillo y claro, el conjunto de recomendaciones debe ser aplicado por los investigadores de ensayos clínicos y las instituciones relacionadas para fomentar la integridad de la investigación sanitaria.
Limitaciones y fortalezasHay varias cuestiones para tener en cuenta en los puntos débiles y fuertes de este estudio de desarrollo de consenso. Definir la integridad de la investigación para determinar el alcance del documento no fue sencillo dado que no existe una definición consensuada3,4, sin embargo, es importante reconocer que no hay controversia en cuanto a su significado. Para utilizar con confianza los resultados de la investigación, se espera que se apliquen las normas éticas y profesionales óptimas para llevar a cabo la investigación e informar sobre sus resultados1. Establecer la integridad de forma restrictiva, centrándose en la falta de honradez una vez publicados los resultados, no permite reconocer que es necesario abordar todo el proceso de investigación desde la propia concepción del estudio34. Nuestro trabajo está sujeto a otras limitaciones, como la posibilidad de que el grupo de consenso pueda considerarse derivado de un muestreo de conveniencia y se arriesgue a un sesgo de selección que podría conducir a resultados particulares, o que no haya incluido todas las perspectivas a pesar de un gran esfuerzo por captar el abanico más amplio posible gracias a la técnica (fig. 1); el tamaño de la muestra de nuestro grupo de expertos fue mayor que la mediana de 22 especialistas incluidos en grupos anteriores para la elaboración de guías35. El método de «bola de nieve» es una técnica de muestreo no probabilístico en la que los miembros actuales del panel seleccionan a los futuros miembros, a diferencia de los métodos de muestreo aleatorio que los seleccionan a partir de listas confeccionadas. Además, todos aquellos expertos que puedan considerarse no incluidos podrán enriquecer nuestro trabajo con sus comentarios tras su publicación36. Las encuestas y votaciones se basaron, por la naturaleza del consenso, en opiniones. No todos los expertos respaldaron todas las recomendaciones (los porcentajes de acuerdo se pueden consultar en la tabla 3). Por ejemplo, a pesar del alto nivel de apoyo general (92,3% de aprobación con un buen nivel de acuerdo entre los expertos en la primera ronda), hubo una fuerte objeción individual al papel de los comités de monitorización en la supervisión de la integridad de los datos (tabla 3, recomendación 26). En otro ejemplo, en el que dos especialistas en estadística discreparon sobre la interpretación de la evidencia subyacente37,38 utilizada para formular la recomendación relativa a la significación estadística (tabla 3, recomendación 33), el nivel global de apoyo apenas superó el umbral de consenso (69,2% de aprobación en la segunda ronda). Para poner en práctica esta recomendación, pueden ser útiles los ejemplos de estrategias analíticas válidas en presencia de múltiples resultados notificados en la literatura publicada39–41. El uso de la revisión paraguas19 añadió amplitud y objetividad42. Por ejemplo, la recomendación relativa a la aportación de los redactores de textos médicos profesionales surgió de una revisión sistemática (tabla 3, recomendación 40)19, no de la aportación de ningún experto.
En relación con el potencial conflicto de intereses de los expertos incluidos, se adjuntaron todas sus declaraciones como material suplementario (Apéndice 1). Otra crítica podría ser que estos hayan sido demasiado indulgentes, inclinándose por promover la integridad de forma suave, en lugar de crear retos para los investigadores, comités, editores, etc. mediante recomendaciones difíciles de aplicar. Al informar explícitamente de los niveles de acuerdo y compartir abiertamente los datos del consenso, pretendíamos maximizar la transparencia para los lectores. No cabe duda de que este documento de consenso deberá actualizarse y revisarse en el futuro.
Nuestro punto fuerte es que capturamos las cuestiones de integridad a lo largo de todo el ciclo de vida de los ECA, avanzando sobre recomendaciones generales anteriores2,3. Utilizando técnicas de consenso establecidas y con base científica20–24, desarrollamos un conjunto de recomendaciones específico que es exhaustivo, metodológicamente reproducible e informado de forma transparente (pueden consultarse los apéndices relativos a las contribuciones de los autores, las declaraciones de conflicto de intereses y el apartado de trasparencia e intercambio de datos). La revisión paraguas19 aportó una elevada proporción de recomendaciones a aquellas proporcionadas por los expertos, que contaban con una amplia y adecuada gama de conocimientos y experiencia, incluida la representación de los pacientes y público general43.
Es importante señalar que los propios expertos no eran autores de ECA con retracciones ni la validez de sus trabajos estaba siendo cuestionada mediante las llamadas «expression of concern» relacionadas con la integridad. Además, somos conscientes de que la ubicación de la reunión final de consenso, El Cairo, puede poner la investigación egipcia en el punto de mira. En este sentido, es importante examinar objetivamente el panorama de las retractaciones. La distribución actual del número de estudios clínicos retractados en la base de datos Retraction Watch44 muestra que EE. UU., Japón y China ocupan los primeros puestos, no Egipto (fig. 4).

Número de estudios clínicos retractados por país según Retraction Watch Database.
Tomado de: http://retractiondatabase.org44.
Este documento de consenso puede ser utilizado por cualquier experto, ya que ofrece orientaciones generales aplicables a la disciplina de investigación de los ECA. A modo de ejemplo explicativo, el hecho de que British Journal of Obstetrics and Gynaecology (BJOG) lleve la palabra British en su nombre y que la revista tenga una base histórica y física dentro del territorio británico no significa que sus artículos publicados solo pertenezcan o tengan implicaciones para las mujeres británicas o la práctica obstétrico-ginecológica británica. De la misma manera, su republicación en la revista de la Sociedad Española de Médicos de Atención Primaria (SEMERGEN) no significa que solo sea aplicable a la medicina de familia. Por lo tanto, no prevemos que esto afecte a la aplicabilidad general de nuestro consenso.
Un miembro del grupo de expertos (NM) tenía experiencia en la representación de pacientes y público en la investigación27, tanto ayudando a los autores de los ECA en el diseño y la realización, como actuando como miembro de comités de supervisión y de evaluación de solicitudes de subvención de ECA para su financiación.
Las respuestas fueron anónimas y los umbrales de aprobación de las recomendaciones se determinaron de forma objetiva y se sometieron a análisis de sensibilidad. En la bibliografía existen varios métodos estadísticos para establecer el grado de consenso entre los encuestados dentro de un panel, como el número estipulado de rondas, el análisis subjetivo, el APMO, la moda, la valoración de media/mediana y otros23. Los parámetros elegidos, el APMO y el umbral arbitrario predefinido se encuentran entre los más utilizados23. Además, se empleó el IQR como medida de dispersión de las respuestas, para cuantificar la fuerza del acuerdo entre los expertos.
El umbral de aprobación se determinó arbitrariamente durante la ronda final de votaciones, algo que debería mejorarse en futuros consensos. A través de varios ciclos de consenso y retroalimentación, cada recomendación se redactó para lograr la máxima claridad de significado y evitar ambigüedades. Con una orientación práctica, el conjunto de afirmaciones ofrece recomendaciones para integrar y mejorar las normas de integridad de los ECA. Todas aquellas incluidas en el conjunto final estuvieron respaldadas por un alto nivel de consenso por parte del grupo multidisciplinar de expertos.
Interpretación de los resultadosNuestro documento de consenso proporciona un conjunto de recomendaciones y principios relativos a la integridad de los ECA para que cada organización pueda elaborar manuales con especificaciones a la hora de participar en ECA y llevarlos a cabo45. Es por ello, que este consenso servirá de base para crear planes de aplicación, manuales, normas y políticas en las instituciones y organizaciones de los diferentes agentes implicados en los ECA que ayuden a promover la integridad de los mismos.
Los investigadores, las instituciones, los organismos y los editores desempeñan funciones complementarias e interconectadas en el mantenimiento de la integridad de los ECA. La colaboración y la armonización son esenciales para hacer frente a las complejidades y obstáculos en el desarrollo de los ECA y existe una iniciativa que ha tratado de desarrollar un documento de procedimiento operativo estándar46 que deberá actualizarse a la luz de nuestras recomendaciones de consenso.
Proteger y promover la integridad de los ECA requiere un enfoque multifactorial, por ejemplo, combinar la formación continua en las mejores prácticas de investigación en ensayos clínicos dirigida a diferentes agentes implicados con mejoras de la gobernanza y la auditoría, la automatización de los controles de integridad en los manuscritos de ECA, o la formación de editores y revisores en metodología. Para todo ello es necesario invertir en la puesta en marcha y mantenimiento de infraestructuras de investigación clínica que respalden la realización de ECA fiables.
Los errores (no) intencionados pueden reducirse, pero no eliminarse por completo. Un aspecto clave de la mejora continua es poder admitir equivocaciones sin riesgo de sentirse señalados47. Para mejorar la credibilidad de los ECA en la investigación sanitaria, se necesitan urgentemente estrategias que reduzcan la probabilidad de errores48, algo en lo que hace hincapié nuestro documento.
En lo que respecta a la supervisión de los ensayos, este trabajo sugiere que los comités de ética, además de su función tradicional de evaluación y aprobación de protocolos antes de que un ensayo pueda comenzar, deberían desempeñar un papel activo en el seguimiento de la realización del mismo. Las deliberaciones de los comités de supervisión del ensayo deberían documentarse formalmente y, en el futuro, es posible que deban hacerse públicas durante el transcurso de este para responder a las crecientes exigencias de transparencia. Una vez finalizado el ensayo, los presidentes de los comités de ética y de supervisión podrían entregar certificados de autenticidad a los autores para que los presenten junto con los manuscritos de sus ECA.
Este documento de consenso reconoce a las revistas biomédicas como agentes clave en la integridad de los ECA, como es obvio por la proporción de editores y revisores representados en nuestro grupo de consenso. Se observó que la mayoría de las instrucciones de las revistas a los autores carecían de detalles suficientes para guiarlos sobre cómo reportar los hallazgos de sus ensayos con integridad49. Se destacó específicamente que este era el caso de la información relacionada con la notificación de la aprobación ética, las fuentes de información, los posibles conflictos de intereses, el registro de los ensayos y los planes de análisis estadístico49–53. En este sentido, también es previsible que las revistas desarrollen e implementen en el futuro controles automatizados de la integridad de los ECA, al igual que han hecho para la detección del plagio54,55.
Cuando se presenta una alegación de posible mala conducta científica, las revistas tienen la obligación de investigar de manera imparcial sobre la gestión de los conflictos de intereses de sus editores, revisores y asesores con una política explícita. Nuestro documento aconseja a los autores que participen activamente en el proceso de investigación que presenten sus datos brutos no identificables para que sean examinados si es necesario. Como cuestión de buena práctica con respecto a la promoción de la transparencia, los autores pueden enviar voluntariamente y de forma electrónica los datos en un repositorio al mismo tiempo que entregan el manuscrito. No hay ninguna razón lógica para no ser proactivos, a la espera de que esto se convierta en un requisito obligatorio, que sin duda es el siguiente paso natural en el desarrollo de la declaración de intercambio de datos del ICMJE56. Esperemos que estas recomendaciones ayuden a reducir el riesgo de reclamaciones.
La prevalencia notificada de mala conducta científica oscila de 2-14%57. Durante una investigación, la mala conducta puede parecer obvia o levantar sospechas cuando, por ejemplo, existen observaciones repetidas (copiar y pegar filas y columnas) o una fórmula para generar datos falsos en una hoja de cálculo. Sin embargo, en todos los casos, antes de tomar una decisión sobre si un ECA es fraudulento, es necesario investigar detenidamente los datos brutos. Las acusaciones erróneas perjudican a la ciencia y a la asistencia sanitaria47,58,59. Por lo tanto, la detección precisa de conductas indebidas debe ser uno de los objetivos de la investigación futura para apoyar la revisión por pares y la evaluación en fases posteriores a la publicación. En la actualidad, la educación en materia de ética, gobernanza y supervisión de la investigación puede ser más eficaz para generar evidencia científica fiable60,61.
ConclusionesLa aplicación de este consenso internacional multidisciplinar contribuirá a mejorar la integridad de los ensayos clínicos.
FinanciaciónEsta investigación no recibió financiación externa. Los gastos de viaje, alojamiento y facilitación logística del consenso corrieron a cargo de Upper Egypt Assisted Reproduction Conference (UEARS) 2022.
Aprobación éticaNo se aplica a este tipo de estudios
ColaboradoresLas funciones de los autores se enumeran en la tabla 1 de acuerdo con la Taxonomía de Funciones de los Colaboradores (CRediT)62.
Conflicto de interesesLos posibles conflictos de intereses de todos los autores figuran en el Apéndice 1.
Intercambio de datosEl 27 de junio de 2022 se publicó una descripción detallada de los resultados de cada encuesta en Open Science Forum en https://osf.io/92ahr.
Republicación del original «International multi-stakeholder consensus statement on clinical trial integrity» del Professor Khalid Khan y el Cairo Consensus Group on Research Integrity (DOI: 10.1111/1471-0528.17451) con el permiso explícito de «John Wiley & Sons Ltd»).
Khalid S Khan es investigador distinguido de la Universidad de Granada financiado por el programa Beatriz Galindo (modalidad senior) del Ministerio de Educación español; M. Núñez-Núñez tiene un contrato de investigación del Instituto de Investigación Carlos III (Juan Rodés JR23/00025).
El grupo de consenso de El Cairo agradece a UEARS su apoyo a esta iniciativa de integridad en la investigación, así como al Comité de Cooperación Científica y Tecnológica (COMSTECH) de un consorcio de 57 países.
