








Remdesivir y nirmatrelvir-ritonavir (NTV/r) son los fármacos antivirales disponibles en España para evitar la progresión de la COVID-19 leve-moderada en la población vulnerable. Los ensayos clínicos pivotales de ambos se realizaron en unas condiciones epidemiológicas diferentes a las actuales. Por tanto, su efecto en el escenario actual es incierto.
Pacientes y métodosSe realizó un estudio observacional multicéntrico retrospectivo de cohortes en 16 servicios de urgencia hospitalaria (SUH) españoles. Se recogieron los datos de todos los pacientes con COVID-19 leve-moderada que consultaron en un SUH durante los 7 primeros días de clínica del 1 de enero al 31 de agosto de 2022. Se evaluó la incidencia de ingreso o muerte por cualquier causa a 30 días (evento combinado) tras el alta del SUH, así como la aparición de reacciones adversas a medicamentos (RAM) graves. Los datos se analizaron mediante la regresión múltiple de Cox y las curvas de supervivencia estandarizadas.
ResultadosSe incluyeron 2.533 pacientes. El uso de NTV/r se asoció con una disminución del riesgo de evento combinado respecto a cuidados habituales (SOC): hazard ratio ajustado (HRa): 0,528; intervalo de confianza al 97,5% (IC 97,5%): 0,330-0,845; número de pacientes a tratar para evitar un evento, 24 (IC 97,5%: 13-283). No se detectó diferencia entre remdesivir y SOC: HRa: 0,835; IC 97,5%: 0,524-1,394. No se detectó ninguna RAM grave.
ConclusiónEl uso precoz de NTV/r se asoció con una menor progresión de la COVID-19 leve-moderada en la población vulnerable, mientras que no se encontraron diferencias entre remdesivir y SOC. Su uso fue seguro.
Remdesivir and nirmatrelvir-ritonavir (NTV/r) are the antiviral drugs available in Spain to prevent progression of mild-moderate COVID-19 in vulnerable populations. The pivotal clinical trials of both were conducted under different epidemiological conditions than the current ones. Therefore, their effect in the current setting is uncertain.
Patients and methodsA retrospective, multicentre, observational cohort study was conducted in 16 Spanish hospital emergency departments (ED). Data were collected from all patients with mild to moderate COVID-19 who presented to an ED in the first seven days after symptom onset between 1st January and 31st August 2022. The incidence of hospitalisation or death from any cause at 30 days (composite endpoint) after discharge from the ED was assessed, as was the occurrence of serious adverse drug reactions (ADRs). Data were analysed using Cox multiple regression and standardised survival curves.
ResultsA total of 2533 patients were included. The use of NTV/r was associated with a reduced risk of the combined endpoint compared to standard of care (SOC): adjusted hazard ratio (aHR) 0.528, 97.5% confidence interval (97.5%CI): 0.330-0.845; number of patients to treat to avoid an event, 24 (97,5%CI 13-283). No difference was detected between remdesivir and SOC: aHR 0.835: 97,5%CI: 0.524-1.394. No serious ADRs were identified.
ConclusionEarly use of NTV/r was associated with less risk of progression of mild to moderate COVID-19 in vulnerable patients, while no differences were found between remdesivir and SOC. Their use was safe.

En el escenario actual, la COVID-19 es una enfermedad que cursa habitualmente de forma benigna, presentando los pacientes una sintomatología seudogripal. Sin embargo, existen factores de riesgo individuales que pueden hacer que la enfermedad progrese a formas graves que condicionen el ingreso hospitalario y la supervivencia de los pacientes1,2. Desde el descubrimiento de la enfermedad, se han llevado a cabo varios ensayos clínicos en la población no vacunada que demostraron la utilidad de diferentes moléculas, administradas de forma precoz durante el curso de la enfermedad, para evitar su progresión en los pacientes con factores de riesgo. En dichos ensayos las variantes virales predominantes fueron Alfa y Delta3–6. Estos tratamientos están disponibles en España desde finales de 2021 y su uso se priorizó para los pacientes con alta vulnerabilidad a través de recomendaciones emitidas por la Agencia Española del Medicamento y Productos Sanitarios (AEMPS). Dichas recomendaciones se fueron ampliando progresivamente conforme hubo mayor disponibilidad de los tratamientos7,8.
Tanto las características de la población como las variantes circulantes han ido cambiando desde que se llevaron a cabo dichos ensayos clínicos. Actualmente, la población española tiene una alta proporción de inmunidad frente al virus, bien a través de la vacunación o de una infección previa9,10. Además, las variantes del SARS-CoV-2 que circulan actualmente pertenecen a diferentes linajes de Ómicron, las cuales difieren de las que circulaban cuando se realizaron los ensayos pivotales de los fármacos11. Esto ha hecho que los anticuerpos monoclonales, como Sotrovimab3, hayan perdido eficacia neutralizante frente a las variantes actuales12. El antiviral de molécula pequeña molnupiravir no mostró eficacia para evitar la muerte o ingreso hospitalario a 28 días en un ensayo clínico reciente13.
A pesar de la disminución de la morbimortalidad atribuida al SARS-CoV-2 tras los sucesivos cambios de variantes virales y las campañas de vacunación masiva14, aún sigue siendo un patógeno peligroso para los pacientes de edad avanzada15, inmunosuprimidos o comórbidos (población vulnerable16), con una mortalidad que dobla a la atribuida a la gripe17–19. Es por ello por lo que siguen siendo necesarios fármacos eficaces que detengan la progresión de la enfermedad y eviten la descompensación de patología crónica subyacente20.
Por último, no se conoce el efecto de remdesivir5 y nirmatrelvir-ritonavir (NTV/r)6 para evitar la progresión de la COVID-19 leve-moderada en la población española con las condiciones actuales.
El objetivo principal del presente estudio es determinar el efecto de remdesivir y NTV/r, comparados cada uno con cuidados habituales, sobre el ingreso o la mortalidad por cualquier causa a 30 días de los pacientes vulnerables con COVID-19 leve-moderada que consultan en un servicio de urgencia hospitalario (SUH) en España. Los objetivos secundarios son determinar este efecto en los pacientes mayores de 65 años, mayores de 80 años, inmunosuprimidos, y en aquellos pacientes que consultaron antes del tercer y quinto día de clínica; así como detectar los eventos adversos graves asociados al tratamiento antiviral.
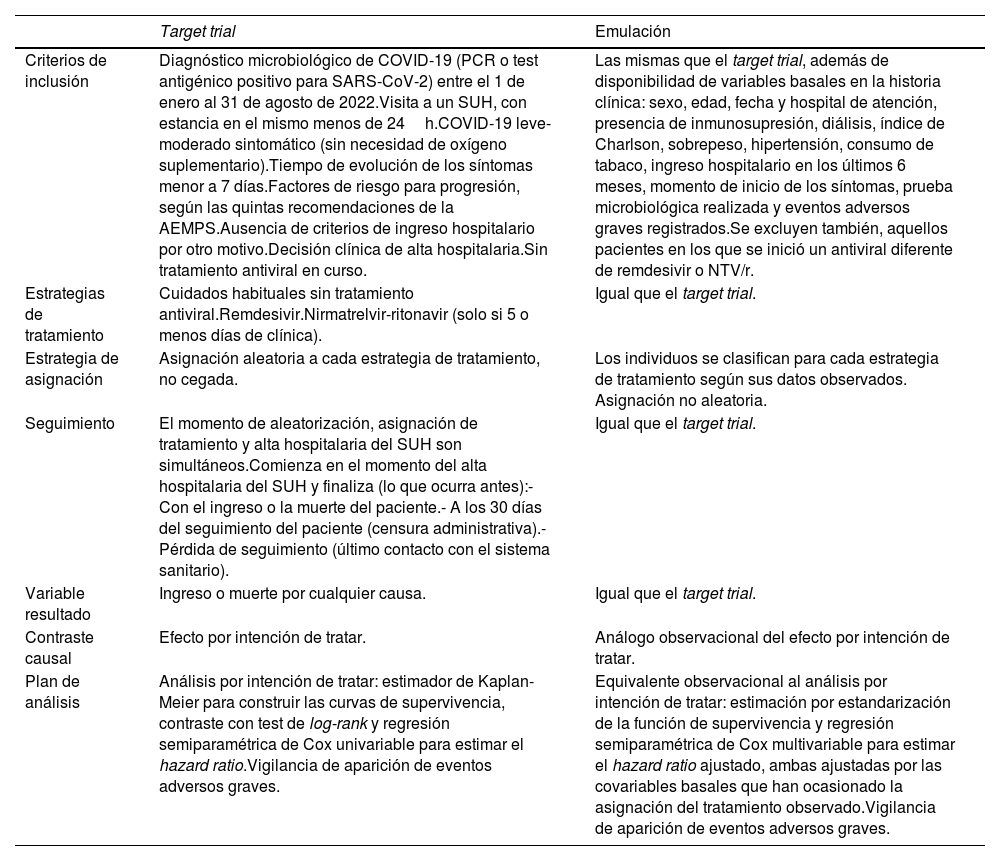
Material y métodosDiseño y pacientesSe realizó un estudio de cohortes retrospectivo multicéntrico en 16 SUH españoles (tabla S1 del material suplementario). Se tuvo en cuenta el marco teórico del target trial emulation (emulación de un ensayo clínico) propuesto por Hernán M et al. para la concepción del proyecto (tabla 1), pues es de gran utilidad cuando la puesta en práctica de un teórico ensayo clínico no es posible por limitación de recursos o inconvenientes éticos y solo se puede extraer evidencia a partir de datos observacionales, tal como ocurre en el presente estudio21,22. Se examinaron las historias clínicas de aquellos pacientes mayores de 18 años con una prueba de detección de infección aguda positiva (test antigénico o reacción en cadena de la polimerasa [PCR]) atendidos en los SUH participantes del 1 de enero al 31 de agosto de 2022. Se incluyeron aquellos pacientes con COVID-19 leve-moderada sintomática (es decir, sin necesidad de oxígeno suplementario), decisión del médico de urgencias de manejo ambulatorio y que fueran candidatos a tratamiento antiviral según las quintas recomendaciones de la AEMPS, publicadas el 2 de agosto de 2022 (disponible en el material suplementario)7. Se excluyeron los registros repetidos, así como los pacientes con duración de la clínica de más de 7 días en el momento de la valoración en el SUH, con COVID-19 grave de inicio, con decisión de ingreso hospitalario por otro motivo en la visita índice, en los que se decidiera iniciar tratamiento con un antiviral diferente de remdesivir o NTV/r, con una estancia en urgencias de más de 24 h, con ausencia de sintomatología atribuible a la infección por SARS-CoV-2 o que ya estuvieran con un tratamiento antiviral en curso previo a su visita al SUH.
Especificación del ensayo clínico objetivo (target trial) y emulación de este (emulation) en el presente estudio
| Target trial | Emulación | |
|---|---|---|
| Criterios de inclusión | Diagnóstico microbiológico de COVID-19 (PCR o test antigénico positivo para SARS-CoV-2) entre el 1 de enero al 31 de agosto de 2022.Visita a un SUH, con estancia en el mismo menos de 24h.COVID-19 leve-moderado sintomático (sin necesidad de oxígeno suplementario).Tiempo de evolución de los síntomas menor a 7 días.Factores de riesgo para progresión, según las quintas recomendaciones de la AEMPS.Ausencia de criterios de ingreso hospitalario por otro motivo.Decisión clínica de alta hospitalaria.Sin tratamiento antiviral en curso. | Las mismas que el target trial, además de disponibilidad de variables basales en la historia clínica: sexo, edad, fecha y hospital de atención, presencia de inmunosupresión, diálisis, índice de Charlson, sobrepeso, hipertensión, consumo de tabaco, ingreso hospitalario en los últimos 6 meses, momento de inicio de los síntomas, prueba microbiológica realizada y eventos adversos graves registrados.Se excluyen también, aquellos pacientes en los que se inició un antiviral diferente de remdesivir o NTV/r. |
| Estrategias de tratamiento | Cuidados habituales sin tratamiento antiviral.Remdesivir.Nirmatrelvir-ritonavir (solo si 5 o menos días de clínica). | Igual que el target trial. |
| Estrategia de asignación | Asignación aleatoria a cada estrategia de tratamiento, no cegada. | Los individuos se clasifican para cada estrategia de tratamiento según sus datos observados. Asignación no aleatoria. |
| Seguimiento | El momento de aleatorización, asignación de tratamiento y alta hospitalaria del SUH son simultáneos.Comienza en el momento del alta hospitalaria del SUH y finaliza (lo que ocurra antes):- Con el ingreso o la muerte del paciente.- A los 30 días del seguimiento del paciente (censura administrativa).- Pérdida de seguimiento (último contacto con el sistema sanitario). | Igual que el target trial. |
| Variable resultado | Ingreso o muerte por cualquier causa. | Igual que el target trial. |
| Contraste causal | Efecto por intención de tratar. | Análogo observacional del efecto por intención de tratar. |
| Plan de análisis | Análisis por intención de tratar: estimador de Kaplan-Meier para construir las curvas de supervivencia, contraste con test de log-rank y regresión semiparamétrica de Cox univariable para estimar el hazard ratio.Vigilancia de aparición de eventos adversos graves. | Equivalente observacional al análisis por intención de tratar: estimación por estandarización de la función de supervivencia y regresión semiparamétrica de Cox multivariable para estimar el hazard ratio ajustado, ambas ajustadas por las covariables basales que han ocasionado la asignación del tratamiento observado.Vigilancia de aparición de eventos adversos graves. |
AEMPS: Agencia Española del Medicamento y Productos Sanitarios; PCR: reacción en cadena de la polimerasa; NTV/r: nirmatrelvir-ritonavir; SUH: servicios de urgencia hospitalaria
Para este estudio se han seguido las recomendaciones de la iniciativa Strengthening the Reporting of Observational Studies in Epidemiology (STROBE)23, cuya lista de verificación puede encontrarse en la tabla S2 del material suplementario. Además, se respetaron los preceptos éticos formulados en la Declaración de Helsinki y se obtuvo el dictamen favorable del CEIM (Comité Ético de Investigación con Medicamentos) del Hospital Clínico San Carlos con el código 22/588-O_M_NoSP. Así mismo, el estudio se inscribió en el Registro Español de Estudios Clínicos con el código 0108-2022-OBS.
Variable principalSe consideró como variable principal el evento combinado de muerte o ingreso a 30 días por cualquier causa. El seguimiento del paciente comenzó en el momento en que se dio el alta del SUH y se vigiló la aparición del evento combinado mediante la revisión de su historia clínica, tanto hospitalaria como de atención primaria.
Otras variablesSe recogieron variables demográficas (sexo, edad, fecha y hospital de atención), clínicas (inmunosupresión según la definición dada por la AEMPS7, uso de tratamiento renal sustitutivo crónico, índice de Charlson, sobrepeso, hipertensión, consumo de tabaco, ingreso hospitalario en el semestre previo, momento de inicio de síntomas atribuibles a la infección por SARS-CoV-2, momento de llegada y alta del SUH), microbiológicas (forma de diagnóstico, variante aislada en caso de haberse realizado secuenciación), estado vacunal, tratamiento recibido (cuidados habituales sin antiviral, remdesivir o NTV/r) y si el paciente ya había padecido con anterioridad COVID-19. También se recogió si el paciente sufrió una reacción adversa grave atribuible al tratamiento antiviral, definida como aquélla que produjera la muerte o riesgo de muerte del paciente, necesidad de ingreso hospitalario, prolongación del ingreso hospitalario, incapacidad o una anomalía fetal. Todos estos datos se recabaron a partir de la historia clínica de cada hospital participante por parte de los investigadores en un formulario electrónico confeccionado con REDCap24, en el periodo del 1 de enero de 2023 al 30 de abril de 2024.
Tamaño muestralEl tamaño muestral se calculó en base al hazard ratio (HR) para la ocurrencia del evento combinado, tomando como referencia los resultados publicados por Arbel R et al.25. En este estudio, se observó una incidencia acumulada a 30 días del 0,44% en la rama del antiviral y del 1,90% en la rama de cuidados habituales (standard of care [SOC]), que resultó en un HR no ajustado de 0,23. Con un nivel de error tipo I del 5%, realizamos un ajuste por multiplicidad dado el análisis planeado de comparar cada antiviral con el SOC, por lo que resultó en el 2,5% para cada comparación, es decir, remdesivir vs. SOC y NTV/r vs SOC. De esta forma, con una potencia del 80%, se estimó un tamaño muestral necesario sin pérdidas de 1.043 pacientes por cada rama. Además, teniendo en consideración unas pérdidas del 15% por censura u otros motivos, el tamaño final estimado necesario por rama fue de 1.228 pacientes (3.684 en total).
Análisis estadísticoSe realizó un análisis descriptivo de la muestra. Las variables categóricas se analizaron a través de su frecuencia absoluta y relativa (porcentaje), y sus distribuciones entre las diferentes ramas de tratamiento se compararon con la prueba Chi-cuadrado. Las variables continuas, con su mediana, primer y tercer cuartiles. Las comparaciones entre las 3 ramas se hicieron con la prueba no paramétrica de Kruskal-Wallis. En cuanto a las variables tiempo hasta evento, se comparó la incidencia de eventos entre ramas mediate la prueba no paramétrica de log-rank.
Se ajustó un modelo explicativo de regresión de Cox para el evento combinado, tomando como variable explicativa principal el tratamiento antiviral recibido (remdesivir, NTV/r o solo cuidados habituales). Se estimó el HR con su intervalo de confianza, tanto crudo (modelo univariable) como ajustado (HRa) por covariables confusoras: sexo, edad, inmunosupresión, ingreso hospitalario en los últimos 6 meses, índice de Charlson, diálisis, hipertensión, sobrepeso, estado vacunal, haber pasado la COVID-19 en el pasado y día de síntomas en que fue atendido el paciente. Las variables continuas no fueron categorizadas. En el modelo multivariable se tomó en cuenta la agrupación de pacientes en diferentes clústers (hospitales) mediante un modelo de fragilidad, para tener en cuenta esta posible fuente de heterogeneidad. Además, con las mismas covariables confusoras, se estimó la diferencia de incidencia acumulada estandarizada entre los antivirales y los cuidados habituales sin antiviral mediante el paquete estadístico stdReg, teniendo también en cuenta la agrupación de pacientes en hospitales como cluster. En ambos análisis (regresión de Cox y estandarización), para evitar que la dependencia de las observaciones procedentes del mismo hospital pudiera conllevar a una subestimación del error estándar, la estimación de la varianza se hizo mediante estimadores robustos tipo sandwich26,27. En caso de registrarse una diferencia de incidencia estandarizada a 30 días cuyo intervalo de confianza no incluyera al cero, se calculó el número necesario para tratar (NNT) o el número necesario para hacer daño (NNH), definidos como el inverso de la diferencia de incidencia acumulada al final del seguimiento28. Estos cálculos se realizaron para la muestra completa, para los pacientes mayores de 65 y 80 años, los pacientes inmunosuprimidos y según los días de síntomas (menos de 3 y 5 días).
Asimismo, se examinó la posible influencia de censura informativa mediante la determinación del HRa e incidencia acumulada a 30 días estandarizada para el objetivo principal en 2 muestras simuladas sin censura, una en la que los pacientes con pérdida de seguimiento no hubieran tenido el evento al final del seguimiento a 30 días (mejor mundo) y otra en la que todos los pacientes censurados hubieran tenido el evento en el momento de la censura (peor mundo)26.
Se fijó el error tipo I en 0,05 y todos los test fueron bilaterales. Dado el ajuste por multiplicidad que se realizó solo para el objetivo principal (cohorte completa), las estimaciones se acompañaron de su intervalo de confianza al 97,5% (IC 97,5%). Para el resto de las estimaciones de los análisis secundarios, se determinó su intervalo de confianza al 95% (IC 95%). Se utilizó el programa R versión 4.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
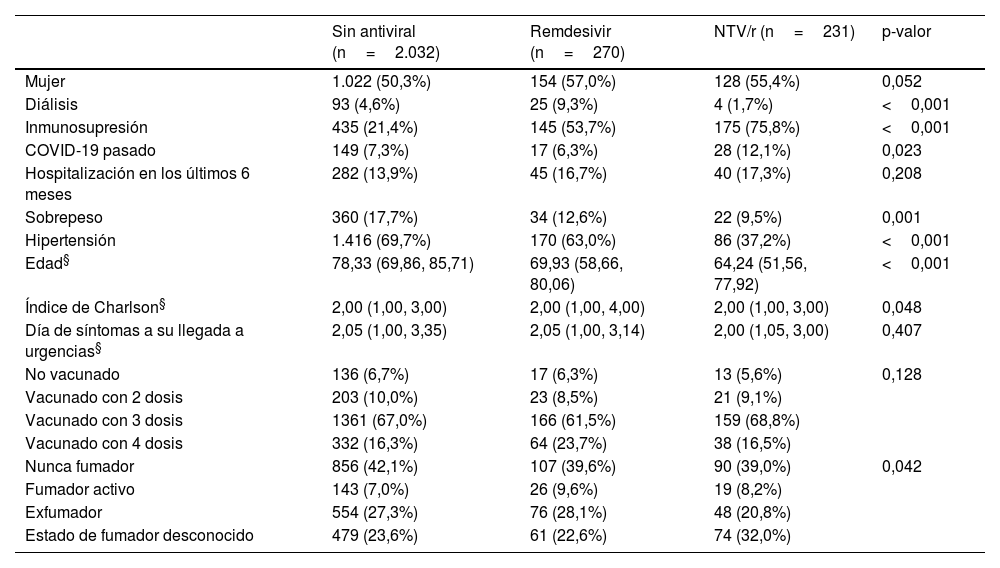
ResultadosDescripción de la muestraEn la figura 1 se muestra el flujograma de inclusión del estudio. Se examinaron 36.729 registros y finalmente se incluyeron 2.533 pacientes. En la tabla 2 se pueden apreciar las características basales de los pacientes en cada rama de tratamiento recibido (cuidados habituales sin antiviral, remdesivir o NTV/r). Se registró una mayor proporción de pacientes inmunosuprimidos en aquellos que recibieron antivirales, mientras que los que no los recibieron eran de mayor edad. Hubo una menor representación de pacientes en diálisis y con sobrepeso en los pacientes que recibieron NTV/r, en tanto que esta rama registró una mayor proporción de episodios considerados como reinfecciones. Fue una cohorte ampliamente vacunada, con una proporción de vacunación completa (3 o más dosis) en todas las ramas de más del 80%. La figura 2 muestra las curvas de incidencia acumulada cruda para cada una de las ramas, así como los pacientes en riesgo en cada momento del seguimiento. La mediana de seguimiento global fue de 30 días (primer y tercer cuartiles, 30-30). En 216 pacientes (8,5%) el seguimiento no fue completo. La rama de NTV/r tuvo menos censura que el resto. En 228 pacientes (9,0%) se registró el evento de interés, de los cuales 23 (0,9%) fueron fallecimientos y 205 (8,1%) ingresos hospitalarios. El diagnóstico microbiológico se hizo por PCR en 978 casos (38,6%), de los que se realizó secuenciación en 279 (28,5% de los pacientes con PCR). Se aisló variante Delta en 26 pacientes (2,7% de los pacientes con PCR), en tanto que en el resto de las secuencias realizadas se determinaron diferentes variantes de Ómicron. No hubo datos faltantes en las covariables basales.

Características basales de la muestra
| Sin antiviral (n=2.032) | Remdesivir (n=270) | NTV/r (n=231) | p-valor | |
|---|---|---|---|---|
| Mujer | 1.022 (50,3%) | 154 (57,0%) | 128 (55,4%) | 0,052 |
| Diálisis | 93 (4,6%) | 25 (9,3%) | 4 (1,7%) | <0,001 |
| Inmunosupresión | 435 (21,4%) | 145 (53,7%) | 175 (75,8%) | <0,001 |
| COVID-19 pasado | 149 (7,3%) | 17 (6,3%) | 28 (12,1%) | 0,023 |
| Hospitalización en los últimos 6 meses | 282 (13,9%) | 45 (16,7%) | 40 (17,3%) | 0,208 |
| Sobrepeso | 360 (17,7%) | 34 (12,6%) | 22 (9,5%) | 0,001 |
| Hipertensión | 1.416 (69,7%) | 170 (63,0%) | 86 (37,2%) | <0,001 |
| Edad§ | 78,33 (69,86, 85,71) | 69,93 (58,66, 80,06) | 64,24 (51,56, 77,92) | <0,001 |
| Índice de Charlson§ | 2,00 (1,00, 3,00) | 2,00 (1,00, 4,00) | 2,00 (1,00, 3,00) | 0,048 |
| Día de síntomas a su llegada a urgencias§ | 2,05 (1,00, 3,35) | 2,05 (1,00, 3,14) | 2,00 (1,05, 3,00) | 0,407 |
| No vacunado | 136 (6,7%) | 17 (6,3%) | 13 (5,6%) | 0,128 |
| Vacunado con 2 dosis | 203 (10,0%) | 23 (8,5%) | 21 (9,1%) | |
| Vacunado con 3 dosis | 1361 (67,0%) | 166 (61,5%) | 159 (68,8%) | |
| Vacunado con 4 dosis | 332 (16,3%) | 64 (23,7%) | 38 (16,5%) | |
| Nunca fumador | 856 (42,1%) | 107 (39,6%) | 90 (39,0%) | 0,042 |
| Fumador activo | 143 (7,0%) | 26 (9,6%) | 19 (8,2%) | |
| Exfumador | 554 (27,3%) | 76 (28,1%) | 48 (20,8%) | |
| Estado de fumador desconocido | 479 (23,6%) | 61 (22,6%) | 74 (32,0%) |
Variables cualitativas expresadas como frecuencia absoluta y relativa entre paréntesis, con p-valor calculado con la prueba de Chi-cuadrado. Variables cuantitativas (§) expresadas como mediana y rango intercuartílico entre paréntesis, con p-valor calculado con la prueba no paramétrica de Kruskal-Wallis.
NTV/r: nirmatrelvir-ritonavir.
.")
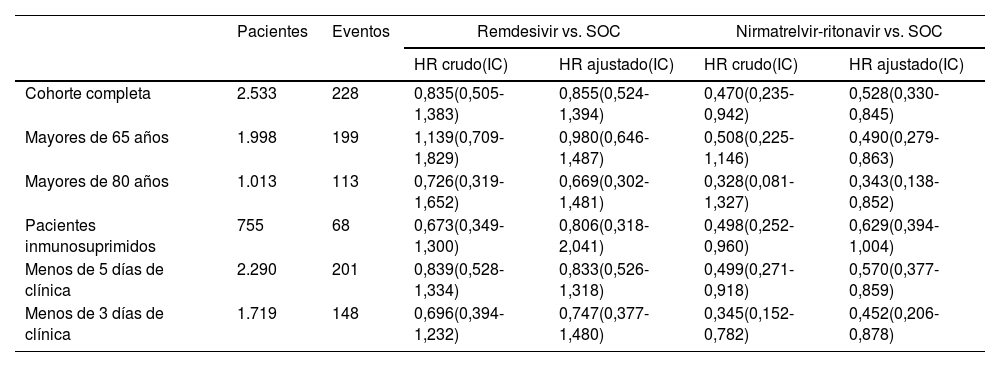
En la tabla 3 y figura 3 se puede apreciar el resultado del análisis para la cohorte completa. Se registró un HRa de 0,528 (IC 97,5%: 0,330-0,845) asociado al tratamiento con NTV/r respecto al SOC. Esta protección se tradujo en una diferencia de incidencia acumulada estandarizada de evento combinado a 30 días respecto a cuidados habituales del 4,3% (IC 97,5%: 0,4-8,3%). No se pudo rechazar la hipótesis nula de igualdad de efecto de remdesivir respecto a SOC: HRa: 0,855 (IC 97,5%: 0,524-1,394), con una diferencia en incidencia acumulada estandarizada del 1,9% (IC 97,5%: −0,3-5,6%).
Resultados de los modelos de regresión de Cox estimados
| Pacientes | Eventos | Remdesivir vs. SOC | Nirmatrelvir-ritonavir vs. SOC | |||
|---|---|---|---|---|---|---|
| HR crudo(IC) | HR ajustado(IC) | HR crudo(IC) | HR ajustado(IC) | |||
| Cohorte completa | 2.533 | 228 | 0,835(0,505-1,383) | 0,855(0,524-1,394) | 0,470(0,235-0,942) | 0,528(0,330-0,845) |
| Mayores de 65 años | 1.998 | 199 | 1,139(0,709-1,829) | 0,980(0,646-1,487) | 0,508(0,225-1,146) | 0,490(0,279-0,863) |
| Mayores de 80 años | 1.013 | 113 | 0,726(0,319-1,652) | 0,669(0,302-1,481) | 0,328(0,081-1,327) | 0,343(0,138-0,852) |
| Pacientes inmunosuprimidos | 755 | 68 | 0,673(0,349-1,300) | 0,806(0,318-2,041) | 0,498(0,252-0,960) | 0,629(0,394-1,004) |
| Menos de 5 días de clínica | 2.290 | 201 | 0,839(0,528-1,334) | 0,833(0,526-1,318) | 0,499(0,271-0,918) | 0,570(0,377-0,859) |
| Menos de 3 días de clínica | 1.719 | 148 | 0,696(0,394-1,232) | 0,747(0,377-1,480) | 0,345(0,152-0,782) | 0,452(0,206-0,878) |
El intervalo de confianza (IC) se proporciona al 97,5% en la cohorte completa, y al 95% para el resto de los análisis. Cada fármaco se compara con cuidados habituales (SOC), sin antiviral.
HR: hazard ratio; vs: versus.

Curvas de diferencia de incidencia acumulada estandarizada de los fármacos comparados con cuidados habituales. En la leyenda figura, junto a la comparación, la disminución de la incidencia acumulada estandarizada a 30 días con su intervalo de confianza al 97,5%. A mayor diferencia positiva, mayor magnitud del efecto protector. NTV/r: nirmatrelvir/ritonavir; REM: remdesivir. Líneas continuas: estimación puntual. Líneas discontinuas: intervalo de confianza al 97,5%.
La tabla 3 y las figuras S1 y S2 del material suplementario muestran el resultado del resto de los análisis realizados. El tratamiento con NTV/r se asoció a un HRa que osciló entre 0,343 y 0,570 dependiendo del subgrupo estudiado, salvo para pacientes inmunosuprimidos, en los que la estimación puntual del efecto protector encontrado (HRa: 0,629) no permitió rechazar la hipótesis nula de igualdad de efecto respecto al SOC (IC 95%: 0,394-1,004). En ninguno de los análisis secundarios con remdesivir se obtuvo una asociación que permitiera detectar diferencias respecto al SOC.
Número necesario de pacientes para tratarLas figuras 3, S1 y S2 del material suplementario muestran las curvas de diferencia de incidencia acumulada estandarizada para cada uno de los fármacos comparados con los cuidados habituales. Esta diferencia incluyó el cero para remdesivir, pero sí se obtuvo un intervalo de confianza estrictamente positivo para NTV/r en la cohorte completa, en los pacientes mayores de 65 años y en aquellos que consultaron antes de 3 y 5 días de clínica. Se puede apreciar así mismo el solapamiento parcial de los intervalos de confianza de ambos fármacos. El NNT para evitar una hospitalización o ingreso a 30 días por cualquier motivo de NTV/r en la población diana fue de 24 pacientes (IC 97,5%: 13-283). En los análisis secundarios para NTV/r, en los pacientes mayores de 65 años fue de 20 (IC 95%: 11-182), en los pacientes con menos de 5 días de síntomas, 27 (IC 95%: 14-538) y en pacientes con menos de 3 días de síntomas, 20 (IC 95%: 10-741). Para el resto de los subgrupos con NTV/r el intervalo de confianza de la diferencia de incidencia acumulada estandarizada incluyó el cero.
Análisis de sensibilidadEn el escenario más optimista (mejor mundo) en el que todos los pacientes con pérdida de seguimiento antes de los 30 días se considera que no tuvieron el evento combinado al final del seguimiento, el HRa fue de 0,854 (IC 97,5%: 0,526-1,386) para remdesivir y de 0,533 (IC 97,5%: 0,331-0,858) para NTV/r. La diferencia en incidencia acumulada a 30 días de remdesivir respecto a SOC fue del 1,3% (IC 97,5%: −3,0-5,5%), y para el NTV/r del 4,2% (IC 97,5%: 0,3-8,0%).
En el escenario más pesimista (peor mundo), en el que se considera que todos los pacientes con censura presentan el evento combinado en el momento de la pérdida de seguimiento, el HRa fue de 0,988 (IC 97,5%: 0,578-1,689) para remdesivir y de 0,439 (IC 97,5%: 0,288-0,670) para NTV/r. La diferencia en incidencia acumulada a 30 días de remdesivir respecto a SOC fue del 0,2% (IC 97,5%: −8,7-9,1%), y para NTV/r del 9,8% (IC 97,5%: 0,9-18,6%).
Eventos adversos graves atribuidos a los antiviralesNo se registró ningún evento adverso grave atribuible a los antivirales.
DiscusiónEn los pacientes con riesgo de progresión de la enfermedad debido a factores de riesgo subyacentes, el tratamiento precoz de la COVID-19 leve-moderada con NTV/r se asoció con una disminución del riesgo de ingreso o muerte por cualquier causa a 30 días. Esta asociación se reprodujo en algunos análisis de subgrupos, si bien estas comparaciones estuvieron limitadas por la disminución que conllevan del tamaño muestral. Esta asociación protectora es consistente pues se ajustó por potenciales variables confusoras mediante regresión múltiple y estandarización y, además, se tuvo en cuenta el factor hospital como posible fuente de heterogeneidad en el efecto. Estos resultados encontrados se han replicado en otras localizaciones geográficas25,29, con una asociación protectora frente al ingreso y muerte similar a la encontrada en este estudio.
En el caso de remdesivir, aunque las estimaciones puntuales sugieren un efecto protector, sus intervalos de confianza incluyeron el uno para la regresión múltiple de Cox y el cero en la diferencia de incidencias estandarizadas, por lo que no es posible rechazar la hipótesis nula de igualdad de efectos entre remdesivir y SOC. Esta diferencia encontrada del efecto de ambos antivirales frente al estándar de cuidados sin antiviral no debe interpretarse en términos de superioridad de un antiviral frente al otro, puesto que el diseño del estudio no permite hacer esta comparación y, además, los intervalos de confianza de las estimaciones de ambos antivirales se solapan. Asimismo, en otro estudio realizado en EE. UU. en adultos ingresados sin necesidad de oxígeno suplementario con un tamaño muestral mayor (11.6376 pacientes, casi 46 veces más respecto a nuestro estudio), se objetivó una asociación protectora de remdesivir frente a la mortalidad a 28 días (aHR: 0,83; IC 95%: 0,76-0,90)30. En otro estudio realizado en México en pacientes inmunosuprimidos con COVID-19 leve-moderada, también se encontró una asociación protectora frente al ingreso y muerte a 28 días (aHR: 0,16; IC 95%: 0,06-0,44)31. La discrepancia de resultados del presente trabajo respecto a lo conocido previamente a través de las mencionadas publicaciones podría estar en relación con el tamaño muestral de nuestro estudio, que no alcanzó la previsión inicial.
Respecto a la seguridad, no se detectó ningún evento adverso grave. Esta información de efectividad y seguridad supone, hasta donde conocemos, una aportación nueva acerca de los resultados clínicos del tratamiento antiviral frente a la COVID-19 leve-moderada en población española con riesgo de progresión que consulta en los SUH.
Nuestro estudio tiene varias fortalezas. En primer lugar, las condiciones de aplicación de los fármacos durante el periodo de reclutamiento son parecidas a las que existen actualmente, tanto en los criterios de prescripción de los antivirales como en cuanto a las características clínico-virológicas, con una población con un alto grado de inmunidad por infecciones previas y vacunación, y con variantes de una virulencia parecida (todas son diferentes variantes Ómicron)8,11,20. Por tanto, es de esperar que los resultados hallados se mantengan en la actualidad. En segundo lugar, la toma en cuenta de la posible heterogeneidad de los pacientes atribuible a su atención en diferentes hospitales hace que los resultados sean más generalizables en el territorio español26. En tercer lugar, la estimación de la cuantificación del efecto no se limita a una tasa relativa de riesgos (HRa), sino que se proporciona una estimación absoluta del mismo en términos de diferencia de incidencia acumulada y NNT. Esta estimación absoluta permite tener una información más completa acerca de la estimación del efecto27,28. Además, los resultados del análisis de sensibilidad para determinar el impacto de una posible censura informativa lo descartan, pues proporcionan unas estimaciones en sentido y magnitud muy parecidas entre ellas y similares a las encontradas en el análisis principal26. Por último, el uso del marco conceptual de emulación de un ensayo clínico proporciona un respaldo sólido al depurar la magnitud del efecto y aproximar una estimación causal21,22.
La principal limitación de este estudio es que la tasa de prescripción de antivirales en los SUH españoles no fue la esperada y no se alcanzó el tamaño muestral exigido para las ramas de pacientes con tratamiento antiviral. Ello se compensó parcialmente con un mayor número de pacientes sin tratamiento antiviral. Esta limitación ocasiona que se pierda potencia estadística para encontrar un posible efecto real subyacente de las intervenciones26. Por otra parte, la naturaleza retrospectiva del estudio hace que sea más vulnerable a la pérdida de información que no estuviera registrada en la historia clínica. Se subsanó con un examen riguroso de las historias clínicas de los pacientes incluidos y un proceso exhaustivo de depuración de la base de datos de cada hospital con los respectivos equipos investigadores. Como consecuencia de esto, no hubo datos faltantes en la base de datos. Si bien no se registraron eventos adversos graves atribuibles a los antivirales, no se puede descartar que aparecieran eventos adversos leves que hayan pasado desapercibidos, tales como la disgeusia, frecuente evento adverso leve y transitorio de NTV/r6. Por último, aunque se ha realizado un esfuerzo considerable en depurar el efecto para intentar asumir causalidad en el análisis, nunca se puede asegurar dicha relación causal en un estudio observacional pues no es posible descartar completamente la presencia de variables confusoras no medidas o la existencia de confusión residual no ajustada por el modelo elegido21. Esta situación, que puede existir en cualquier estudio de naturaleza observacional, hace que la incertidumbre pueda ser mayor a la medida y que las estimaciones puntuales puedan verse afectadas32. En cualquier caso, en el presente estudio se ha llegado a la misma conclusión en las estimaciones a través de 2 métodos de ajuste diferentes, regresión múltiple y estandarización, por lo que podemos afirmar que los hallazgos son robustos.
Es necesario realizar nuevos estudios en población española con un tamaño muestral adecuado para investigar el efecto de remdesivir en la población actual, así como la efectividad en población inmunosuprimida para ambos antivirales. Respecto a esta población, es de vital interés distinguir entre diferentes grados y tipos de inmunosupresión (humoral, celular, etc.) para poder individualizar y optimizar las pautas de tratamiento33.
ConclusiónEl tratamiento precoz con NTV/r de la COVID-19 leve-moderada en los pacientes con riesgo de progresión en SUH españoles se asoció con una disminución del riesgo de ingreso y muerte por cualquier causa a 30 días. No se halló una diferencia en el efecto de remdesivir respecto al SOC, si bien la potencia del estudio fue subóptima pues no se pudo alcanzar el tamaño muestral planeado. Ambos tratamientos fueron muy seguros para los pacientes pues no se registró ningún evento adverso grave.
FinanciaciónEste trabajo ha sido financiado parcialmente por la beca de investigación PID2022-137050NB-I00. Ministerio de Ciencia e Innovación, España (TPP). Se trató de una colaboración no condicionada, por lo que ello no ha influido en el diseño, recogida de datos, análisis de estos ni en la redacción o revisión del manuscrito.
Los gastos de publicación del artículo han sido financiados por la Fundación para la Investigación e Innovación Biomédica del Hospital Universitario Infanta Sofía y Hospital Universitario del Henares (FIIB HUIS HHEN).
Consideraciones éticasSe ha llevado a cabo de conformidad con el Código de Ética de la Asociación Médica Mundial (Declaración de Helsinky). Se obtuvo la aprobación del CEIm (Comité Ético de Investigación con Medicamentos) del Hospital Universitario Clínico San Carlos de Madrid, con el código 22/588-O_M_NoSP. Así mismo, el estudio se inscribió en el Registro Español de Estudios Clínicos con el código 0108-2022-OBS. Se consideró exento de la solicitud de consentimiento informado a los participantes.
Conflicto de interesesCMRL ha recibido honorarios en concepto de conferencias y financiación de asistencia a congresos por parte de Gilead y Pfizer. MSRG ha recibido financiación para la asistencia a congresos por parte de Gilead. El resto de los autores declaran no tener conflicto de intereses en relación con el presente artículo.
A Miguel Ángel Luque Fernández por sus consejos en el abordaje metodológico.
recomendados
Medicina Clínica Práctica


Medicina Clínica sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas