










Cholestatic liver diseases comprise a heterogeneous group of disorders affecting both adult and pediatric population, characterized by alterations in bile formation, secretion, or flow, leading to the accumulation of bile acids and other toxic substances in the liver. In recent years, advances in new pharmacological therapies, the availability of next-generation genetic sequencing techniques, and the development of specific treatments for genetic cholestasis have transformed the diagnostic and therapeutic approach to these conditions. This document, jointly prepared by the “Asociación Española para el Estudio del Hígado” (AEEH) and the “Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica” (SEGHNP), presents a national evidence-based guideline for the diagnosis and management of hepatic cholestasis in Spain. It addresses recommendations for differential diagnosis, diagnostic algorithms, indications for genetic studies, treatment and follow-up criteria in diseases such as primary biliary cholangitis, primary sclerosing cholangitis, genetic cholestasis, intrahepatic cholestasis of pregnancy, and vanishing bile duct syndrome. In addition, recommendations are included for the management of extrahepatic complications, indications for liver transplantation, and special considerations in pregnancy and childhood. The guideline emphasizes the importance of a multidisciplinary approach, the use of non-invasive tools for risk stratification, and the incorporation of new targeted therapies, with the aim of improving the prognosis and quality of life of patients affected by cholestatic liver diseases.
Las enfermedades colestásicas hepáticas comprenden un grupo heterogéneo de trastornos que afectan tanto a adultos como a población pediátrica, caracterizados por alteraciones en la formación, secreción o flujo biliar, lo que conlleva a la acumulación de ácidos biliares y otras sustancias tóxicas en el hígado. En los últimos años, el avance en nuevas terapias farmacológicas, la disponibilidad de técnicas de secuenciación genética de nueva generación y el desarrollo de tratamientos específicos para colestasis genética han transformado el abordaje diagnóstico y terapéutico de estas patologías. Este documento, elaborado conjuntamente por la Asociación Española para el Estudio del Hígado (AEEH) y la Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP), presenta una guía nacional basada en la evidencia para el diagnóstico y manejo de la colestasis hepática en España. Se abordan recomendaciones para el diagnóstico diferencial, algoritmos diagnósticos, indicaciones de estudio genético, criterios de tratamiento y seguimiento en patologías como la colangitis biliar primaria, colangitis esclerosante primaria, colestasis genética, colestasis intrahepática del embarazo y el síndrome de desaparición de conductos biliares. Además, se incluyen recomendaciones sobre el manejo de complicaciones extrahepáticas, indicaciones de trasplante hepático y consideraciones especiales en el embarazo y la infancia. La guía enfatiza la importancia de un enfoque multidisciplinar, la utilización de herramientas no invasivas para la estratificación del riesgo y la incorporación de nuevas terapias dirigidas, con el objetivo de mejorar el pronóstico y la calidad de vida de los pacientes afectados por enfermedades colestásicas hepáticas.
Cholestasis encompasses a spectrum of hepatic disorders characterized by impaired bile formation, secretion, or flow, resulting in the accumulation of bile acids (BA) and other toxic substances in the liver. These conditions can affect both pediatric and adult populations. While each condition presents distinct pathophysiological mechanisms, they often share clinical manifestations, such as pruritus, jaundice, and progressive liver damage that may culminate in cirrhosis or necessitate liver transplantation (LT) over time.
In recent years, several pivotal developments have underscored the necessity of standardizing the diagnosis and management of cholestatic diseases: (1) the approval and availability of new pharmacological treatments for primary biliary cholangitis (PBC),1,2 (2) relevant information for the management of primary sclerosing cholangitis (PSC) has been published,3 (3) the advancement and widespread accessibility of next-generation sequencing (NGS) techniques, which have enhanced the diagnosis of genetic cholestatic diseases in both pediatric and adult populations,4 and (4) the availability of therapeutic agents for the management of genetic cholestasis.5,6
This document will focus on providing guidance for the diagnosis and treatment of PBC, PSC (adults and children), genetic cholestasis (adults and children), cholestasis of pregnancy, and vanishing bile duct syndrome in Spain.
MethodologyIn 2023, the Asociación Española para el Estudio del Hígado (AEEH) and the Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP) convened a panel of experts to develop national clinical practice guidelines for the diagnosis and management of cholestatic liver diseases in Spain. The guideline development followed internationally recognized standard operating procedures.
The expert panel first identified key clinical questions for each topic, structured using the PICO format (P: patient or population; I: intervention; C: comparison; O: outcome). These questions underwent a Delphi process involving 30 pediatric and adult hepatologists. Sufficient consensus was achieved after two rounds of voting.
Following this, a systematic review of the literature was conducted using PubMed. Each panelist was responsible for drafting statements and recommendations within a specific section of the guidelines, which were then reviewed and discussed by the full group. The panel met virtually five times, and all recommendations were collectively debated and approved (Supplementary Table 1).
The level of evidence supporting each statement was graded according to the Oxford Centre for Evidence-Based Medicine system, and the strength of each recommendation was classified as either ‘strong’ or ‘weak’.7 The final endorsement of all recommendations was obtained through a second Delphi round.
CholestasisHow can cholestasis be diagnosed in adults?Recommendations:
- •
In case of cholestasis, detailed history and physical examination is mandatory. (LoE 3, strong recommendation).
- •
The abdominal ultrasound should be the first test to perform in case of cholestasis. (LoE 3, strong recommendation).
- •
AMA and PBC-specific ANA should be screened in patients with unexplained cholestasis. (LoE 3, strong recommendation).
- •
MRCP should be performed if previous tests are negative. (LoE 3, strong recommendation).
- •
Liver biopsy should be considered in patients with chronic cholestasis who remain undiagnosed. (LoE 3, strong recommendation).
- •
Genetic testing is recommended when previous tests are negative. (LoE 4, strong recommendation).
- •
In selected cases with clinical history indicating a potential genetic cause of cholestasis, genetic testing can be performed before the liver biopsy. (LoE 5, weak recommendation).
Cholestasis can be asymptomatic or manifest as fatigue, pruritus, right upper quadrant abdominal discomfort, and jaundice. Biochemical markers include increased serum alkaline phosphatase (ALP) and gamma-glutamyl transpeptidase (GGT) levels, followed by conjugated hyperbilirubinemia at more advanced stages. Cholestasis is considered chronic if it lasts for >6 months and can be classified as intrahepatic or extrahepatic. Chronic cholestatic liver diseases often have an asymptomatic course over months or years and can be identified by elevated serum ALP levels. However, ALP can also originate from the bone, intestine, and placenta, and isolated ALP elevation should prompt the investigation of extrahepatic causes of this abnormality, including bone disease, vitamin D deficiency, or pregnancy.8
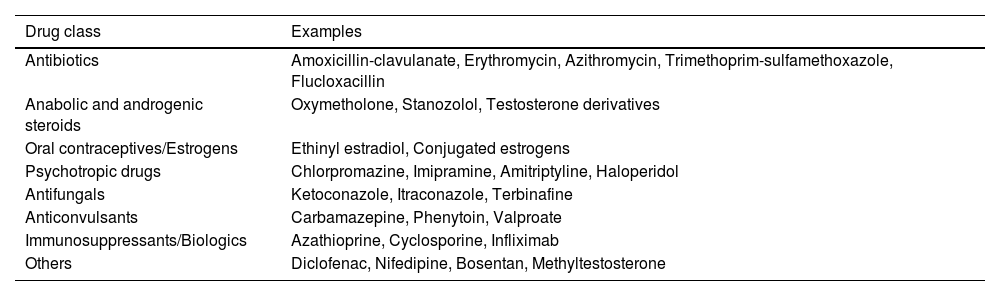
Elevation of ALP and GGT levels indicates a hepatic source, and patients need to undergo a structured diagnostic algorithm to establish the cause of this abnormality. A complete clinical history should include the presence of symptoms (pruritus, jaundice, abdominal pain), concomitant diseases (including thyroid disease, celiac disease, sicca syndrome, systemic sclerosis, inflammatory bowel disease [IBD]), drug or herbal product consumption (Table 1), previous history of trauma or immunotherapy, family history of biliary problems or autoimmune diseases, alcohol consumption, and drug abuse. Physical examination should include the search for jaundice, xanthelasma, scratch lesions, hepatomegaly, splenomegaly, and other signs of chronic liver disease. Initial diagnostic testing should include abdominal ultrasound to rule out an extrahepatic cause of cholestasis and antimitochondrial antibodies (AMA) and specific antinuclear antibodies (ANA) to confirm the diagnosis of PBC (see below). If negative, extended imaging with magnetic resonance cholangiopancreatography (MRCP)3 can be performed to study the morphology of the intrahepatic and extrahepatic biliary trees. In case of normality of the previous tests, literature recommends performing a liver biopsy to detect diseases involving the intrahepatic bile ducts (PBC, small-duct PSC), infiltrative or granulomatous liver diseases, nodular regenerative hyperplasia, peliosis, or signs of portosinusoidal vascular disease (PSVD) and hepatocellular cholestasis. Finally, recently published guidelines recommend performing a genetic test to discard the presence of genetic cholestasis when previously mentioned tests are negative.4 However, in cases of suspected genetic cholestasis, where liver biopsy information is limited, it is advisable to consider genetic testing prior to conducting a liver biopsy. This is particularly pertinent in selected cases where clinical indicators suggest potential genetic cholestasis, such as pruritus beginning in childhood or early adulthood, pruritus during pregnancy, the presence of biliary stones, a history of cholecystectomy, or a relevant family history (Fig. 1).
Drugs related to cholestatic drug-induced liver injury.
| Drug class | Examples |
|---|---|
| Antibiotics | Amoxicillin-clavulanate, Erythromycin, Azithromycin, Trimethoprim-sulfamethoxazole, Flucloxacillin |
| Anabolic and androgenic steroids | Oxymetholone, Stanozolol, Testosterone derivatives |
| Oral contraceptives/Estrogens | Ethinyl estradiol, Conjugated estrogens |
| Psychotropic drugs | Chlorpromazine, Imipramine, Amitriptyline, Haloperidol |
| Antifungals | Ketoconazole, Itraconazole, Terbinafine |
| Anticonvulsants | Carbamazepine, Phenytoin, Valproate |
| Immunosuppressants/Biologics | Azathioprine, Cyclosporine, Infliximab |
| Others | Diclofenac, Nifedipine, Bosentan, Methyltestosterone |
.")
Evaluation of adult patients with cholestasis. Determination of BA may be considered in patients without conclusive findings in prior testing and with normal MRI/MRCP results. If BA are elevated, a genetic study may be considered without prior histological evaluation. ** A liver biopsy should be performed if not previously done (in cases where a genetic study was done without prior biopsy).
Recommendations:
- •
Prolonged neonatal jaundice, defined as jaundice beyond two weeks of age in both breast- and formula-fed infants, must be investigated, even in an otherwise “well-appearing” neonate. (LoE 5, strong recommendation).
- •
If cholestasis is present (defined as serum direct or conjugated bilirubin>1mg/dl if total bilirubin<5mg/dl or >20% of total bilirubin if it is ≥5mg/dl) the infant must be immediately referred to a specialized pediatric liver disease center. (LoE 5, strong recommendation).
- •
In the setting of cholestasis, treatment with nutritional support and fat-soluble vitamins should be initiated promptly. (LoE 4, strong recommendation).
- •
Ultrasound must be performed as an initial assessment of cholestasis to exclude biliary obstruction. (LoE 5, strong recommendation).
- •
An elevated international normalized ratio (INR) without response to parenteral vitamin K administration should prompt ruling out causes of neonatal acute liver failure. (LoE 5, strong recommendation).
- •
Genetic tests should be performed early for the diagnosis of cholestatic infants. (LoE 5, strong recommendation).
Jaundice is very common in the neonate, especially in breastfed infants, and it is usually secondary to unconjugated hyperbilirubinemia. Cholestatic jaundice is an uncommon condition, occurring in approximately 1 in every 2500 term infants; however, it represents a potentially severe disease. It may go unnoticed because of its temporal overlap with the more common non-cholestatic neonatal jaundice. Cholestasis in infancy is defined as a serum conjugated bilirubin level of >1mg/dl, exceeding 20% of the total bilirubin concentration.
To ensure early detection of cholestasis, prolonged jaundice (defined as jaundice persisting beyond two weeks of age in a breastfed infant or at 2 weeks of age in a formula-fed infant) must be investigated, even in an otherwise well-appearing neonate.9–14 The initial step in the diagnostic approach should be obtaining a blood sample to measure total and conjugated bilirubin, GGT, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and INR, along with urine culture.9–11,15 Stool color should also be assessed, as acholic or pale stools are considered a red flag.9–11 Direct visualization of the stool in the diaper by the physician is recommended as it is not uncommon for parents to fail to recognize acholic or hypocholic stools as abnormal.9–11 If cholestasis is identified, the infant must be referred immediately to a specialized pediatric liver disease center.9–11,14
The priority should be the detection of potential acute life-threatening conditions (e.g., sepsis, acute liver failure, urinary tract infection) or treatable diseases (e.g., inborn errors of metabolism such as galactosemia, hormone deficiency, inborn errors of primary bile acid synthesis) and early diagnosis of biliary atresia.12,14 It is also important to promptly start nutritional support12,15 and fat-soluble vitamin treatment to avoid the severe consequences of its deficiency.16
If prolonged prothrombin time or elevated INR are detected, parenteral vitamin K must be administered. If there is no response to this treatment, the causes of neonatal acute liver failure9 such as gestational alloimmune liver disease (GALD), inborn errors of metabolism, and infections, should be ruled out. GGT levels, whether elevated or within the normal range, provide an important initial clue for guiding stepwise evaluation of cholestatic infants. To further assess potential biliary obstruction, abdominal ultrasound is recommended as the first-line imaging modality in the initial workup for cholestasis.9,12
Advances in genetic testing and understanding of the molecular mechanisms underlying bile formation and secretion have enhanced diagnostic capabilities, particularly in the setting of progressive familial intrahepatic cholestasis (PFIC). Consequently, genetic testing using next-generation sequencing (NGS) should be incorporated earlier in the diagnostic algorithm for infants with cholestasis.14,16–21 NGS has enabled a diagnosis in 22–61% of children with cholestasis,17,19–21 and this variability largely reflects differences in the study populations in which NGS was applied.9,11–13 Larger deletions, duplications, or inversions may not be detected by NGS, and in such cases, additional genetic studies such as whole exome sequencing (WES) or whole genome sequencing (WGS) may be required. It is important to note that variants of unknown significance (VUS) should be interpreted with caution and should not be used as the sole basis for establishing a molecular diagnosis.
Among the different causes of cholestasis in infancy, biliary atresia is the most common cause of pediatric end-stage liver disease and the leading indication for pediatric LT.10 Early diagnosis followed by timely surgical intervention with Kasai hepatic portoenterostomy is crucial and has been associated with improved transplant-free survival.
What is the differential diagnosis for neonatal or infant cholestasis with normal GGT levels?Recommendations:
- •
In infants with cholestasis and normal or low GGT levels, inborn errors in bile acid synthesis, panhypopituitarism, or PFIC should be investigated. (LoE 4, strong recommendation).
- •
Normal GGT levels, elevated serum bile acid levels, and severe pruritus should prompt a diagnosis of PFIC. (LoE 3, strong recommendation).
- •
Recurrent hypoglycemia, cholestasis, micropenis, and septooptic dysplasia should increase the diagnosis of congenital hypopituitarism. (LoE 5, strong recommendation).
In infants with cholestasis, normal or disproportionately low GGT values relative to the degree of cholestasis should prompt investigation to rule out specific conditions. These include inborn errors in primary bile acid synthesis,12,22 panhypopituitarism23,24 and PFIC with normal GGT levels.12,22 In fact, GGT-normal cholestasis beginning in the first months of life, accompanied by elevated serum BA and severe pruritus, should raise clinical suspicion of PFIC. PFIC encompasses a group of rare and heterogeneous autosomal recessive disorders caused by defects in the proteins involved in bile formation. Currently, the diagnosis of these conditions relies primarily on genetic analyses. Mutations in genes, such as ATP8B1, ABCB11, FXR, TJP2, MYO5B, USP53, UNC45, and VPS33B/VIPAS, are associated with cholestasis with normal GGT levels.15,22 In this context, the absence of pruritus and low serum bile acid levels may help differentiate inborn errors in primary bile acid synthesis25,26 from PFIC with normal GGT levels. Conversely, the combination of recurrent episodes of hypoglycemia and cholestasis should guide investigations to rule out congenital hypopituitarism.23,24 The presence of clinical features such as micropenis and septo-optic dysplasia further supports this diagnosis.
How should neonatal or infant cholestasis with elevated GGT level be studied?Recommendations:
- •
Biliary atresia should be ruled out in infants with cholestasis and elevated GGT levels. (LoE 3, strong recommendation).
- •
Loss of color or the presence of acholic stools should be assessed in infants with cholestasis and elevated GGT levels. (LoE 5, strong recommendation).
- •
Liver ultrasound should be performed as a first step in the evaluation of neonatal cholestasis with high GGT levels. (LoE 5, strong recommendation).
- •
There is no single diagnostic test for biliary atresia. Surgical exploration with intraoperative cholangiography is mandatory for disease confirmation. (LoE 3, strong recommendation).
In the diagnostic evaluation of infants with high GGT cholestasis, ruling out biliary atresia must be a priority.10,12,13 Early diagnosis of biliary atresia and early Kasai hepatic portoenterostomy surgery are associated with better transplant-free survival.12,15 In this regard, liver ultrasonography must be performed as a first step to rule out biliary obstructive causes of cholestasis, such as sludge, stones, or choledochal malformations. It also allows for the identification of features suggestive of biliary atresia such as the “triangular cord sign” or an absent/abnormal gallbladder.9,10,12–15 Since no single diagnostic test can definitively confirm biliary atresia, surgical exploration with intraoperative cholangiography is essential to establish the diagnosis.10,12,13,15 A diagnosis of biliary atresia should be suspected in neonates or young infants who present with cholestasis with elevated GGT (>150–200IU/l) and acholic stools or progressive color discoloration. These findings should raise immediate concerns, as stool color is a critical early warning sign. Liver biopsy may be useful in the evaluation of infants with cholestasis10,12,13,15; however, its use should never delay exploratory surgery when there is a high clinical suspicion of biliary atresia. Typical histological findings of biliary atresia include ductal proliferation, bile plugs, and portal fibrosis.10,12,13,15 These changes may be absent in early biopsies (<1 month of age) and may be present in other causes of neonatal cholestasis.10,13–15 Therefore, the differential diagnosis should include biliary atresia, Alagille syndrome, alpha-1 antitrypsin deficiency, cystic fibrosis, MDR3 deficiency, Niemann–Pick type C, neonatal sclerosing cholangitis (e.g., Claudin-1 or DCDC2 deficiency), and other ciliopathies.
How should cholestasis with elevated GGT levels be studied at the onset of childhood or adolescence?Recommendations:
- •
A complete medical history, physical examination, and liver ultrasound should be performed for every patient with cholestasis and elevated GGT levels. (LoE 5, strong recommendation).
- •
Viral, autoimmune, metabolic, and genetic diseases should be included in the differential diagnoses. (LoE 5, strong recommendation).
- •
Liver biopsy may be considered in the absence of these findings. (LoE 5, weak recommendation).
A comprehensive medical assessment is crucial, beginning with a detailed medical history, including family history, travel history, exposure to toxins or medications, presence of concurrent autoimmune diseases, and presence of abdominal pain. It is critical to determine whether the presentation of symptoms suggests acute or chronic onset. Physical examination should be meticulously conducted to identify signs indicative of chronic liver disease, which will guide further diagnostic workup and prompt the consideration of extrahepatic manifestations, such as a cardiac murmur or distinctive facial features associated with Alagille syndrome.
As for neonates, abdominal ultrasound is an essential diagnostic tool for children or adolescents exhibiting high GGT cholestasis, serving to exclude biliary obstruction due to gallstones or biliary sludge, neoplastic growth, or choledochal cysts. Concurrently, the measurement of prothrombin time in cases of high GGT cholestasis is critical for early detection of acute liver failure.
Once biliary obstruction has been excluded, further work-up for liver disease should be guided by physical examination findings. This typically includes blood tests, such as viral serologies or polymerase chain reaction (PCR) assays for pathogens, including Epstein–Barr virus, cytomegalovirus (CMV), hepatitis A, B, C, E, adenovirus, enterovirus, and herpes virus. Additional tests should include the measurement of alpha-1-antitrypsin levels and genotyping if levels are reduced, as well as autoimmunity markers, such as ANA, liver-kidney microsome type 1 (LKM-1) antibodies, and smooth muscle antibodies (SMA). The assessment of ceruloplasmin levels is also recommended.
In certain cases, liver biopsy and genetic sequencing may be indispensable to establish a definitive diagnosis. The differential diagnosis should include conditions such as Alagille syndrome, PSC, secondary sclerosing cholangitis, MDR3 deficiency, and ciliopathies such as DCDC2 or KIF12 deficiencies.
Primary biliary cholangitisHow is PBC diagnosed?Recommendation:
The diagnosis of PBC should be established when two of the following three criteria are met: (a) biochemical evidence of cholestasis based on ALP elevation; (b) presence of AMA or other PBC-specific autoantibodies, including sp100 or gp210; and (c) histological evidence of nonsuppurative destructive cholangitis and destruction of interlobular bile ducts. (LoE 3, strong recommendation)
PBC must be considered in patients with persistent cholestasis, particularly when there is a sustained elevation of serum ALP levels. The cornerstone of PBC diagnosis is the detection of autoantibodies, particularly AMA, which target the E2 subunit of the pyruvate dehydrogenase complex. More than 90% of patients with PBC are positive for AMA. Immunofluorescence (at a titer of at least 1:40) and immunoenzymatically assays are highly specific for the disease in the presence of cholestasis. Additionally, ANA are detected in approximately 30% of patients with PBC. A thorough analysis of ANA is crucial, as certain ANA subtypes are highly specific for PBC (95%), but have relatively low sensitivity. In AMA-negative patients, the presence of ANA-immunofluorescence staining patterns, such as nuclear dots (indicative of anti-sp100 reactivity) or perinuclear rims (suggestive of anti-gp210 reactivity), is diagnostically valuable for confirming PBC.
In addition to ALP levels, other biochemical features are often observed in patients with PBC, although they are not exclusive to this condition. For example, elevated immunoglobulin levels, particularly IgM, are commonly observed. Additionally, these patients may present with elevated serum levels of AST and/or ALT, even in the absence of autoimmune hepatitis (AIH), owing to the extent of liver inflammation and necrosis associated with PBC.
While liver biopsy is not essential for diagnosing PBC because of the high specificity of the autoantibodies in the presence of cholestasis, it has become a valuable diagnostic tool in cases where PBC-specific antibodies are absent or when there is a suspicion of concurrent AIH. Upon examination, the most common finding is chronic, non-suppurative inflammation that leads to the destruction of the interlobular and septal bile ducts, a condition referred to as ‘florid duct lesions’.
Is it necessary to determine the presence of anti-HK1 and anti-KLHL12 antibodies in patients with suspected PBC and other negative antibodies?Recommendation:
- •
Assessment of anti-HK1 and anti-KL12 antibodies is recommended in patients with suspected PBC when conventional autoantibodies are negative, (LoE 3, weak recommendation)
Among the various diagnostic challenges in PBC, identifying AMA-negative patients is one of the most significant, as it represents only approximately 5–10% of cases. In this situation, other autoantibodies can aid in the diagnostic process. In this context, specific ANA associated with PBC, such as sp100 or gp210, can be identified through indirect immunofluorescence in approximately 30% of cases that are negative for AMA.27 More recently, evidence suggests that anti-HK1 and anti-KLHL12 antibodies are present in approximately 40% of patients with PBC who are negative for AMA.28 Additionally, although further studies are needed, anti-HK1-positive individuals have been associated with significantly reduced transplant-free survival (TFS) and a shorter time to liver decompensation.29 Therefore, beyond their diagnostic ability, assessing these antibodies can add valuable prognostic information for patients suspected of having PBC, but who test negative for conventional antibodies.
What is the risk of developing PBC in patients with positive AMA (or specific ANA) without cholestasis?Recommendation:
- •
Although the risk is low, the 5-year incidence rate of PBC is 16% among patients with normal ALP levels and no evidence of advanced liver disease. (Statement, consensus 87%).
Although the presence of AMA (>1/40) is a strong indicator of PBC, AMA alone is not sufficient to establish a definitive diagnosis.30 In the general population, approximately 0.64% of people show AMA positivity,31 although most of these individuals will never develop PBC over time. A French prospective study showed that only one out of six patients (16%) with AMA positivity and normal ALP levels developed PBC within five years.32 More recently, a Chinese retrospective study suggested that this figure could be even lower, with a cumulative 5-year incidence rate of 4.2%.33 Therefore, it seems reasonable to conduct annual liver biochemistry monitoring in patients with positive AMAs and normal liver tests.
Is the determination of IgM levels useful in the diagnosis of PBC?Recommendations:
- •
Serum IgM levels can be measured as a complementary diagnostic parameter in patients with suspected PBC, especially in those who are antibody-negative or have normal serum ALP despite PBC-specific antibody positivity. (LoE 4, weak recommendation).
Elevated IgM levels are commonly observed during the diagnostic workup for PBC.34 While an increase in serum IgM concentration is indicative of polyclonal activity, it is not consistently observed as it is absent in approximately one-third of cases. Consequently, it cannot be regarded as a specific marker for PBC.35 Elevated IgM concentrations can be found in PBC as well as in acute viral infections, lymphomas, and protozoal parasitic infections.36 Thus, although high IgM concentrations are not considered a standard criterion for PBC,35,37 they can support diagnostic suspicion of PBC, particularly in patients with atypical presentations, such as those who are AMA-negative.35,38 It is important to highlight that the diagnostic performance of autoantibodies in PBC varies significantly across populations. One study revealed that a large proportion of Latin American/Hispanic patients with PBC may be AMA-negative. In these cases, IgM positivity has been identified as a potential diagnostic tool for AMA-negative PBC.39 Serum IgM levels may also be an important clinical indicator of PBC in patients with seropositive AMA but normal ALP levels. Ding D et al. performed a retrospective cohort study that included 115 AMA positive patients and normal ALP who were treatment-naïve and who underwent liver biopsy to establish a definitive diagnosis.40 The study showed that baseline serum IgM (>0.733×ULN) and age (>42years) are potential indicators for the diagnosis of PBC.41 Sun et al. studied 169 patients with positive AMA and normal ALP levels and found that serum IgM levels were elevated in 53.3% of cases. Liver biopsies were performed on 67 patients, revealing that 55 (82.1%) exhibited varying degrees of cholangitis activity, consistent with a PBC diagnosis.42
Is it necessary to perform liver biopsy in patients with suspected PBC and negative antibodies?Recommendations:
- •
Liver biopsy is recommended to establish the diagnosis of PBC in cases where specific autoantibodies are negative, particularly in AMA-negative patients, lacking access to PBC-specific ANA antibodies, or when biochemical characteristics of chronic cholestasis are present and PBC is suspected. (LoE 2, strong recommendation).
- •
Histological findings should be interpreted with caution, as characteristic histological lesions may be absent in the early stages of PBC. (LoE 3, strong recommendation).
Liver biopsy is not routinely recommended in patients who meet the noninvasive criteria for PBC diagnosis but should be performed if biochemical and/or serological data are equivocal.37,38,43 Terziroli et al. studied a Swiss PBC cohort that included 30 patients with normal ALP level and positive AMA and/or PBC-specific ANA. Twenty-four (80%) had histological features of PBC, including three AMA-negative and PBC-specific ANA-positive patients. However, most of these patients had elevated GGT and the results should be interpreted with caution. The decision to perform a biopsy in these cases should be individualized based on the clinical context, additional diagnostic indicators, and thorough discussion of risks and benefits to the patient.44
Liver biopsy must be offered as a diagnostic measure in the absence of PBC-specific autoantibodies, unusual biochemical presentation (isolated increase in GGT), suspicion of a PBC/AIH variant, or any suspected hepatic comorbidity (including metabolic associated steatotic liver disease [MASLD] or alcohol-related liver disease).35,37,38,43
Histological examinations for the diagnosis of PBC, as with all liver biopsy interpretations, depend on the size of the liver tissue and the number of analyzable portal tracts.45 Histological alterations in PBC are heterogeneously distributed throughout the liver; therefore, the probability of identifying the presence of cholangitis and bile duct destruction increases with the number of portal tracts present. Thus, a biopsy of adequate quality should include at least 11 portal tracts.35,37,38,43,45 Cytokeratin 7 immunostaining, which highlights the loss of bile ducts, is recommended for bile duct identification.35,38 Hallmarks of PBC include destructive granulomatous lymphocyte cholangitis affecting the interlobular and septal bile ducts, leading to progressive bile duct loss, chronic cholestasis, fibrosis and cirrhosis.38 Other features that may be identified include interface hepatitis, parenchymal necroinflammation, and nodular regenerative hyperplasia.38 Characteristic features may be absent in the early stages of the disease (false-negative biopsies are likely in the very early stages).35,38 The typical diagnostic PBC florid duct lesion with segmental degeneration of the bile ducts and formation of poorly defined non-caseating epithelioid granulomas is found in only a small number of cases.46 However, in autoantibody-negative PBC, the differential diagnosis should include small-duct PSC, sarcoidosis, and other granulomatous diseases such as idiopathic ductopenia, drug-induced liver injury, graft-versus-host disease, and genetic cholestasis.
Liver biopsy remains the gold standard for staging liver fibrosis in patients with chronic liver disease. However, owing to its invasiveness and potential sampling errors, it is not routinely recommended for staging purposes at diagnosis,37,38,43,47 as the stage of PBC can be accurately assessed noninvasively. Histological liver examination for the initial evaluation of disease severity is only recommended in cases of failure, uncertainty or inconsistencies in liver stiffness measurement (LSM) results.35
How is the assessment of the risk of progression performed in patients with PBC?Recommendations:
- •
The stratification of future risk of progression should be based primarily on noninvasive tools, including demographics, clinical manifestations, biochemical and serological profiles, serum markers of fibrosis, and LSM. It can also be based on the results of invasive methods such as histological characteristics, endoscopic examinations, and direct measurements of the hepatic venous pressure gradient (HVPG). (LoE2, strong recommendation).
The clinical course of PBC varies; some patients have slowly progressive disease, while others develop advanced fibrosis and cirrhosis within years and risk liver-related complications and death.43 This risk should be evaluated in all patients. The purpose of evaluating progression risk in PBC patients is: (1) identifying patients at highest risk of adverse outcomes to adapt treatment and monitoring; (2) evaluating therapeutic response to UDCA treatment to determine need for second-line treatment; and (3) detecting early complications, particularly cirrhosis, portal hypertension (PH), and hepatocellular carcinoma (HCC).35 The stratification of future progression risk must be based primarily on noninvasive tools, including demographics, clinical manifestations, biochemical and serological profiles, serum markers of fibrosis, and LSM.9,11,12,18,35,37,38,43 It can also be based on results of invasive methods, such as histological features, endoscopic examinations, and direct measurement of the HVPG.35,38,43
Demographic factors have limited validation as prognostic tools for risk stratification in PBC. Age and sex influence treatment response and long-term outcomes of PBC patients.43 Patients presenting younger (<45 years) are often symptomatic and predisposed to severe progressive disease (rapid ductopenic form in young women) and poorer UDCA response.43 Male sex is associated with later diagnosis, more advanced disease at presentation, poorer UDCA response, and higher HCC risk.35,43 Whether sex serves as an independent prognostic factor for PBC outcome remains questionable. Cheung et al. performed a longitudinal retrospective study of 4355 patients in the Global PBC Study Cohort, including 17 centers across Europe and North America. They evaluated sex and age effects on UDCA treatment response and TFS in PBC patients. Younger patients (≤45 years) had higher serum transaminases than older patients and increased risk of treatment failure, LT, and death. However, in multivariate analysis, sex was not independently associated with response or TFS.48 Nevertheless, Abdulkarim et al. hypothesized that PBC diagnosis in male patients is delayed and prognosis impaired. They performed a case-control study comparing clinical features of 49 male patients and 98 age-matched female patients with PBC. Male patients reported fewer symptoms and were diagnosed at more advanced stages. HCC was exclusively observed in three male patients, with no cases among female patients. They suggested that men had a higher risk of HCC.49
In terms of clinical manifestations, fatigue or pruritus affects more than 50% of patients with PBC during the disease.35 Advanced disease is more likely to be associated with symptoms, and an impaired quality of life which may predict a worse response to UDCA and prognosis, but the evidence is controversial and limited.35,43 Nevertheless, a premature ductopenic variant characterized by severe pruritus, progressive icteric cholestasis, and a limited response to UDCA has been identified. In these patients, histological examination revealed bile duct loss without significant fibrosis, often necessitating LT.43
The biochemical measurements necessary for the initial assessment of disease severity should include a minimum of the following parameters: total and conjugated bilirubin, albumin, platelet count, and ALP and transaminase levels.35 Serum bilirubin has been recognized as the main predictor of poor outcome in PBC.43,50 Baseline bilirubin and albumin values have robust validation and applicability for stratifying risk in PBC.37,38,43 In fact, baseline bilirubin and albumin can stratify UDCA-treated patients into low- (normal bilirubin and albumin), medium- (abnormal bilirubin or albumin), and high-risk (both abnormal bilirubin and albumin) groups. Nevertheless, as both bilirubin and albumin levels are altered only in the advanced stages of the disease, they are not effective markers for risk stratification in patients at early stages of the disease.43 Very high ALP levels at diagnosis are usually indicative of severe and symptomatic disease with a lower likelihood of response to treatment.50 In fact, a study by Carbone et al. with data from the UK-PBC cohort found that elevated ALP and bilirubin at diagnosis, as well as low transaminase levels, younger age, longer time from diagnosis to first line treatment initiation, and worsening ALP from diagnosis were significantly associated with poor treatment response. Interestingly these variables serve to create the UDCA response score (URS) that can be used at diagnosis to define higher-risk patient.51 Gatselis et al. on behalf of the Global PBC Study Group,52 studied the factors associated with progression and outcomes of early stage PBC. From a cohort of 1615 patients with early stage PBC (based on normal levels of albumin and bilirubin) collected data from healthcare evaluations on progression to moderate PBC or advanced-stage PBC. The median follow-up time was 7.9 years, during which 904 patients progressed to a moderate stage, 201 developed advanced disease, and 236 experienced a clinical event. Variables associated with the transition from early to moderate stage included baseline levels of bilirubin, albumin, ALP, transaminase levels, platelet count, and UDCA treatment.52
Regarding serological markers, AMA titers at diagnosis do not correlate with disease stage, severity, or prognosis.43 In contrast, PBC-specific ANA are more frequently detected in patients with high risk PBC, and their presence may be predictive of an unfavorable course.35,37,38,43 Anti-centromere antibodies have also been reported to have a potential prognostic impact, as they are associated with a higher risk of PT.43 Nevertheless, their utility in risk stratification for PBC remains limited due to insufficient validation. Haldar et al. retrospectively analyzed data from 499 patients with PBC at a UK Liver Center for evaluating their diagnostic utility and prognostic significance in terms of TFS. Anti-gp210 autoantibodies were significatively associated with elevated serum transaminases, bilirubin and liver stiffness at presentation, non-response to UDCA and reduced TFS.53 In another retrospective study, Takano et al. showed that IgM levels were associated with cirrhosis-related symptoms and liver-related events, with significantly lower cumulative survival rates in patients with IgM levels ≥240mg/dl.54
When evaluating fibrosis using non-invasive approaches, the AST-to-platelet ratio index (APRI) has been validated in different cohorts as a predictor of adverse events, independent and additive to UDCA response. In a study conducted by Trivedi et al.,55 an APRI of >0.54 at baseline was found to predict the probability of LT or death. Moreover, an elevated APRI at baseline and/or during treatment was associated with an increased risk of adverse events during follow-up, independently of the biochemical response to UDCA.55 LSM, assessed using vibration-controlled transient elastography (TE), is a valuable tool for evaluating prognosis. Among the various alternative liver elastography techniques currently available, TE remains one of the most widely used and recognized worldwide. In an international multicenter retrospective follow-up study of 3985 PBC patients, Corpechot et al.56 identified LSM cut-offs of 8kPa and 15kPa to stratify patients into low, medium and high-risk categories. Patients in the medium- and high-risk categories exhibited approximately 4-fold and 16-fold higher risks of poor clinical outcomes, respectively, compared to those in the low-risk group. Therefore, this study validated LSM as a major independent predictor of PBC clinical outcomes.56 In addition, the 15kPa threshold is also in line with the Baveno VII recommendations, which suggests using >15kPa as the threshold above which compensated advanced chronic liver disease should be strongly suspected, regardless of etiology.57 Other tools for the evaluation of liver stiffness, including real-time tissue elastography (RTE), shear wave elastography (SWE), and magnetic resonance elastography (MRE), have demonstrated excellent diagnostic performance in detecting severe fibrosis and cirrhosis. However, published data evaluating these tools in PBC remain limited.58–61
Beyond TE, prognosis can also be estimated using specific PBC continuous scoring systems. GLOBE score developed by an international multicenter meta-analysis involving 4119 patients with PBC treated with UDCA. The authors identified that age, serum bilirubin, albumin, ALP, and platelet count were independently associated with death or LT. Patients with risk scores>0.30 had significantly shorter TFS compared to matched healthy individuals.62 Another score is the UK-PBC risk score, which was developed using data from a large cohort of patients treated with UDCA. This score, which includes baseline albumin and platelet count, along with bilirubin, transaminases, and ALP levels after 12 months of UDCA therapy, estimates the likelihood of LT or liver-related death within 5, 10, or 15 years. The prognostic performance of both the UK-PBC and GLOBE scores has been validated across different populations in different studies, demonstrating their usefulness as indicators of disease prognosis irrespective of race or ethnicity. In fact, these scores have proven to be better predictors of future cirrhosis-related complications than the classical UDCA response criteria.36
What is the benefit of first-line treatment for patients with PBC? What is the optimal drug and dose of first-line treatment?Recommendations:
- •
Adequate treatment with UDCA in PBC patients has been associated with biochemical improvement and higher TFS. (Statement, consensus 95.6%).
- •
First-line therapy with UDCA at dose of 13–15mg/kg/day should be initiated as soon as the diagnosis of PBC is established. (LoE 1, strong recommendation).
The benefit of UDCA was shown for the first time in a landmark controlled clinical trial by Poupon et al. in 1991, where UDCA treatment for two years showed biochemical improvement and a decrease in IgM in patients with PBC.63 During the open-label phase, patients who received UDCA continuously throughout the entire period experienced slower liver disease progression and reduced need for LT.64 After 10 years of follow-up, the entire cohort demonstrated a higher TFS rate than predicted by the Mayo model. In non-cirrhotic patients, the observed survival was comparable to that of the standardized French population, whereas in cirrhotic patients, it was significantly lower.65 In a US-based randomized clinical trial, patients with PBC receiving UDCA demonstrated better liver-related outcomes and TFS than those receiving placebo. In contrast, a Greek RCT showed no differences in the rates of liver decompensation, liver-related death, or LT.66,67
Since then, UDCA has been extensively studied for its effect on survival and disease progression in patients with PBC. Multiple analyses have highlighted its protective effect against LT and death, particularly when administered at optimal doses and at the early stages of the disease. A comprehensive retrospective analysis of a large registry demonstrated that UDCA had a protective effect against LT or death across all patient groups, except for Caucasian women with an AST/ALT ratio≥1.1.68 Several other studies have supported the role of UDCA in improving the TFS. A meta-analysis of three trials conducted in France, the United States, and Canada demonstrated improved survival in moderate-to-severe PBC cases, particularly in patients with histological stage 4 disease.69,70 Furthermore, a recent multinational cohort study using inverse probability treatment weighting (IPTW) found that UDCA treatment was associated with better TFS across all biochemical stages, independent of dose or GLOBE score. This association was stronger in patients who received a dose ≥13mg/kg/day.71
Biochemical response to UDCA is a well-established predictor of clinical outcomes in patients with PBC. In this regard, a retrospective analysis showed that patients achieving a biochemical response after one year (defined as a 40% reduction in ALP from baseline) had a survival rate comparable to that of a matched Spanish control population, whereas non-responders exhibited suboptimal survival, identifying them as candidates for additional treatment.72 Similarly, smaller retrospective studies confirmed better outcomes in patients who met the Paris-1 biochemical response criteria.73
Larger cohort studies reinforce the clinical benefit of UDCA, as a study from the Global PBC cohort, which included 3902 patients with a median follow-up of 7.8 years, demonstrated that UDCA significantly reduced the risk of LT or death. The overall hazard ratio (HR) was 0.46, leading to a five-year TFS rate of 81%. The benefits were consistent between cirrhotic and non-cirrhotic patients. Additionally, the effect of UDCA varies based on baseline ALP levels, highlighting the importance of early initiation of UDCA treatment to maximize its efficacy and improve patient adherence.74 The survival benefit of UDCA was further supported by a multicenter retrospective analysis of 526 patients from seven Swedish hospitals. This study found that UDCA responders, as defined by the Paris I criteria, had lower mortality and transplantation rates than non-responders or untreated individuals. However, given the retrospective nature of this study, caution is required when interpreting causality.75
Paris I was further refined into the Paris II criteria. In a study of 165 patients with early-stage PBC, the authors identified that patients whose levels of ALP and AST were less than 1.5 times the ULN, and who had normal bilirubin, experienced no adverse liver-related events over a 7-year follow-up, making these markers strong indicators of favorable prognosis. In contrast, all adverse events such as cirrhosis progression or transplantation occurred in non-responders. Compared to previous response models, the Paris II criteria showed the highest specificity and positive predictive value in early PBC, distinguishing low-risk patients.76 Another retrospective study, focusing on histology progression, showed that 80% of patients who failed to achieve an ALP level below 1.67×ULN after two years of treatment progressed in fibrosis stage. Furthermore, ductopenia observed in more than 50% of portal tracts on initial biopsy was strongly associated with both poor treatment response and fibrosis advancement.77
Finally, in terms of dosing, different studies have examined the optimal UDCA dose range. PBC patients randomized to receive doses between 5 and 20mg/kg/day showed lower benefits at doses below 13mg/kg/day, whereas doses exceeding 15mg/kg/day did not provide additional advantages. Consequently, the preferred dosage range is 13–15mg/kg/day.78
Should normalization of blood tests (ALP, bilirubin, AST) be sought after one year of UDCA treatment in patients with PBC? When should the treatment response be evaluated?Recommendations:
- •
Biochemistry normalization is the ideal endpoint treatment for PBC, but it is currently achievable in less than 20–25% at 1 year of treatment. (Statement, consensus 95.6%).
- •
Biochemical response in PBC should be assessed following 12 months of UDCA treatment. (LoE 3, strong recommendation).
- •
Treatment response can be evaluated earlier (after 1–6 months) in high-risk patients. (LoE 4, weak recommendation).
The achievement of normalization of biochemical markers (deep response), particularly ALP and bilirubin (<0.6×ULN), is associated with improved clinical outcomes and reduced progression of liver disease.79 In addition, ALP normalization alone has also been associated with the best survival outcomes. However, attaining these goals may be challenging and not feasible for all patients. Therefore, prioritizing these targets in high-risk groups, such as younger patients (under 62 years) and those with a LSM≥10kPa, is recommended.79 The advantage of achieving normalization of ALP is particularly significant in this population, as its normalization was associated with an absolute increase of 52.8 months in 10-year complication-free survival compared to those who achieved treatment response according to Paris II criteria.
In general, the assessment of treatment response in PBC should occur one year following the initiation of UDCA. Several categorical criteria have been used to evaluate response to UDCA, including Rochester,80,81 Barcelona,72 Paris I and II,76,82 Rotterdam,83 and Toronto77 criteria (Table 2). Although these response systems were developed and validated in diverse clinical settings, they offer a binary classification of treatment response, distinguishing between responders and non-responders. According to these criteria, approximately 40% of patients are classified as non-responders. On the other hand, there are continuous scoring systems, specifically the GLOBE and the UK-PBC score, described previously, which allow for a more refined prognostic stratification. Unlike categorical criteria, these models integrate both biochemical parameters and clinical variables, assessed at baseline and 12 months after treatment initiation. In routine clinical practice, however, the Paris II criteria are the most used, due to their simplicity, intuitive application, and demonstrated better fit for early-stage patients, who currently represent the majority of newly diagnosed PBC cases.
Treatment response criteria in PBC.
| Response criteria | Treatment response |
|---|---|
| Rochester | ALP<2× ULN or Mayo<4.5 |
| Barcelona | Decrease in ALP>40% and ALP<ULN |
| Rotterdam | Bilirubin<ULN and albumin>LLN |
| Torontoa | ALP<1.67× ULN |
| Paris I | ALP<3× ULN, AST<2× ULN and Bilirubin<1Ymg/dl |
| Paris II | ALP<1.5× ULN, AST<1.5× ULN and Bilirubin<1Ymg/dl |
| POISE | ALP<1.67× ULN, reduction of ALP>15% from baseline and bilirubin<ULN |
| GLOBE | Baseline: Age. 12 months: Bilirubin, ALP, albumin, and platelet count |
| UK-PBC | Baseline: Albumin and platelet count at baseline. 12 months: Bilirubin, ALP and AST (or ALT) |
Although treatment response is generally recommended to be assessed at 12 months after initiating UDCA, early evaluation may be particularly beneficial for optimizing management and improving long-term outcomes in high-risk patients. In a retrospective analysis of the Global PBC cohort, an ALP threshold of 1.9×ULN at 6 months of UDCA initiation identified 90% of non-responders according to POISE criteria,84 suggesting that these patients may be candidates for an early initiation of second line drugs.
A new criterion for evaluating UDCA responses at 1 month (ALP≤2.5× ULN and AST≤2× ULN, and total bilirubin ≤1× ULN, Xi’an criteria) predicted the 5-year adverse outcome-free survival rate of UDCA responders versus non-responders (97% vs. 64%). The accuracy of the Xi’an criteria in distinguishing high-risk patients was confirmed in both early- and late-stage PBC85; however, the usefulness of these new criteria must be validated.
What is the treatment for patients with PBC who do not respond to first-line therapy?Recommendation:
- •
Elafibranor, seladelpar and bezafibrate (off-label) can be used in patients with insufficient response or intolerance to UDCA (LoE 1, strong recommendation).
Following the withdrawal of obeticholic acid (OCA) from the European market, three drugs remain available for the management of patients with PBC who exhibit an insufficient response to or intolerance of UDCA: bezafibrate, elafibranor, and seladelpar. These three agents act as agonists of peroxisome proliferator-activated receptors (PPARs), a family of nuclear receptors that play a crucial role in regulating bile acid homeostasis by inhibiting bile acid synthesis, promoting bile acid secretion, reducing bile acid toxicity, and facilitating bile acid detoxification.
Bezafibrate, a pan-PPAR agonist, was evaluated for efficacy in the BEZURSO trial,86 a multicenter, phase 3 clinical trial that randomly assigned 100 patients with inadequate response to UDCA to receive either bezafibrate at a daily dose of 400mg (50 patients) or placebo (50 patients). After 24 months of treatment, 31% of patients in the bezafibrate group achieved complete biochemical response, defined as normalization of bilirubin, ALP, aminotransferases, albumin, and prothrombin time, compared to 0% in the placebo group, and 67% achieved ALP normalization. In terms of safety and tolerability, two patients in each group experienced complications related to end-stage liver disease. Creatinine levels increased by 5% from baseline in the bezafibrate group, whereas it decreased by 3% in the placebo group. Myalgia was reported in 20% of patients in the bezafibrate group and in 10% of those in the placebo group. However, the body of evidence supporting the use of bezafibrate extends beyond the BEZURSO trial. In this context, a retrospective study from Japan demonstrated that the addition of bezafibrate to UDCA significantly improved long-term prognosis by reducing all-cause and liver-related mortality and the necessity for LT.87 Despite these promising outcomes, bezafibrate is not approved for the management of PBC, and its use for this indication remains off-label.
Recently, elafibranor, a dual α and δ PPAR agonist, has been approved as a second-line therapy for PBC based on the findings of the ELATIVE trial.1 In this study, 161 patients were randomized to receive either 80mg of elafibranor once daily or placebo. A treatment response, defined as ALP level<1.67× ULN with a reduction of at least 15% from baseline and normal total bilirubin levels, was observed in 51% of patients receiving elafibranor compared to 4% in the placebo group after 52 weeks of treatment. ALP normalization was achieved in 15% of patients in the elafibranor group, whereas no patients in the placebo group reached this endpoint. A key secondary endpoint of the trial was the change in pruritus through week 24 and 52, assessed using the Worst Itch Numerical Rating Scale (WI-NRS) in patients with moderate-to-severe baseline pruritus. This endpoint was not met, as elafibranor did not lead to a statistically significant improvement in pruritus in the WI-NRS scale. In terms of safety and tolerability, elafibranor was associated with mild adverse effects (96% of any adverse event emerging during treatment period vs. 91% in the placebo group), including abdominal pain, diarrhea, nausea, and vomiting. Serious adverse events occurring during the treatment period were reported in 10% of patients in the elafibranor arm, compared to 13% in the placebo arm.
The efficacy of seladelpar, a selective PPAR δ agonist, was evaluated in the RESPONSE trial.2 This phase 3 clinical trial involved the randomization of 193 patients to receive either 10mg of seladelpar daily or placebo in a 2:1 ratio. A biochemical response, based on the same criteria used in the ELATIVE trial, was achieved by 62% of patients on seladelpar and 20% on placebo. ALP normalization occurred in 25% of patients treated with seladelpar, while none in the placebo group achieved this endpoint. A key secondary endpoint was the change from baseline in the weekly mean pruritus NRS score at month 6 among patients with moderate-to-severe pruritus (NRS≥4). Seladelpar significantly reduced pruritus compared to placebo. In terms of safety and tolerability, adverse events were reported in 86.7% of patients in the seladelpar group and 84.6% in the placebo group, with serious adverse events occurring in 7.0% and 6.2% of patients, respectively.
Currently, there are no head-to-head studies comparing the efficacy and safety of the 3 alternatives, and therefore it is not possible to recommend one drug over the other (Fig. 2). Real world data are needed to determine the best patient profile for each drug. Patients previously treated with OCA or bezafibrate can be treated with one of the new PPAR agonists (similar efficacy and safety as those without a previous second line treatment) but the evidence is very limited and comes from a sub-analysis of the clinical trials presented in abstract form.88
 at 6 months have a low probability of achieving a POISE response at one year. In such cases, the early initiation of a second-line therapy may be considered on an individual basis. * Bezafibrate is not approved for the indication of PBC at the time of writing this document.")
Algorithm for the management of patients with PBC. Response evaluation at one year of treatment can be performed using different validated response criteria, with the most commonly used being the Paris II and POISE criteria. Patients presenting an ALP level greater than 1.9 times the upper limit of normal (× ULN) at 6 months have a low probability of achieving a POISE response at one year. In such cases, the early initiation of a second-line therapy may be considered on an individual basis. * Bezafibrate is not approved for the indication of PBC at the time of writing this document.
In a press release issued in August 2025, a phase IIb/III clinical trial revealed that saroglitazar achieved the primary composite endpoint, exhibiting a 48.5% treatment difference in biochemical response compared to placebo (p<0.001).89 At the time of these guidelines’ publication, no further trial data were available.
The addition of OCA and bezafibrate to UDCA (also known as triple therapy) has shown excellent results in patients with insufficient response to UDCA in the preliminary report of a randomized controlled trial conducted in several countries around the world with ALP normalization and biochemical remission rates at 24 weeks of treatment of 61.1% and 66.7%,90 respectively. In addition, real world observational studies have also shown good results of the triple therapy in patients who failed second line therapy to OCA or bezafibrate. However, after revocation of OCA commercialization in Europe, the use of this combination in real-world clinical practice is rapidly declining, as OCA is no longer being supplied to treatment centers except in exceptional cases individually evaluated by the Spanish Medicines Agency.
Should patients with PBC-related cirrhosis be screened for HCC?Recommendation:
- •
Ultrasound screening for HCC every 6 months is recommended for patients with PBC when cirrhosis is established. (LoE 3, strong recommendation).
Patients with PBC are at risk of developing HCC and this risk increases as histological damage progresses.91,92 The estimated incidence of HCC in patients with PBC is 0.36 per 100 patient-years and is higher in men and in patients with advanced disease, at a rate of 3.4 cases per 1000 patient-years.43,93 In addition to male sex, diabetes mellitus, HBV infection, and lack of response to UDCA also increase the risk of developing HCC.93–96 Suboptimal response to UDCA is also a significant risk factor, although UDCA treatment and response do not prevent this risk in patients with cirrhosis. Furthermore, risk stratification is very important, as male non-responders to UDCA without advanced disease have a higher risk of developing HCC than women with UDCA-responsive cirrhosis.93 Therefore, regular HCC screening with ultrasound evaluation every six months is recommended, especially in men and patients with cirrhosis.92,97,9899–104 In male patients without cirrhosis, the recommendation for semiannual HCC surveillance may be considered on an individual basis.
Which patients with PBC should be screened for esophagogastric varices?Recommendation:
- •
Screening for esophagogastric varices using upper gastrointestinal endoscopy is recommended for patients with PBC and established cirrhosis. (LoE 1, strong recommendation).
- •
When cirrhosis is not clearly established, LSM, spleen stiffness measurement and platelet count can help in determining the need for esophagogastric varices screening. (LoE 3, strong recommendation).
PH can develop as a result of established biliary cirrhosis or, in the pre-cirrhotic phase, in conjunction with nodular regenerative hyperplasia. It often manifests during the early stages of the disease.8,46 However, approximately one-third of patients with advanced disease develop varices within a mean of 5.6 years. In this regard, patients who respond to UDCA exhibit an improvement in PH.105 The AASLD,106 EASL guidelines,107 and the different Baveno Consensus meetings57,108 provide recommendations for the management of esophagogastric varices and variceal hemorrhage in cirrhotic patients, which are also applicable to individuals with PBC.109 Moreover, different efforts have been made to identify noninvasive markers predictive of the presence of esophagogastric varices,110–113 underscoring the need for upper gastrointestinal endoscopy for variceal detection. Current strategies rely on platelet counts and TE as criteria for endoscopic surveillance.114 Specifically, the Baveno VI and VII guidelines recommend incorporating criteria, such as LSM>20kPa or platelet counts<150,000/mm3. However, it is important to note that patients with PBC were underrepresented in these studies.38 In addition, a recently published study, found that the presence of cholestasis defined by the presence of an ALP>1.5× ULN increases the possibility of missing high-risk varices by the Baveno and expanded Baveno criteria115 complicating even more the applicability of these guidelines in PBC patients. In some cases, spleen stiffness values greater than 40kPa may also be useful as a cut-off point to indicate upper gastrointestinal endoscopy.
It is important to emphasize that PH in PBC can develop early in the course of the disease, signaling an increased risk of hepatic decompensation and mortality. Consequently, risk stratification is key to identifying patients who may benefit from screening endoscopy.116 Furthermore, beyond its role in assessing the risk of gastrointestinal bleeding, the detection of esophagogastric varices may have additional prognostic significance even in the absence of cirrhosis, as some studies suggest an increased risk of HCC in patients with PBC and varices, further underscoring the importance of early identification and screening.117
When is LT indicated for patients with PBC?Recommendations:
- •
LT should be considered for patients with PBC who meet standard criteria, such as decompensation and/or HCC. Additionally, LT is indicated for those exhibiting poor prognostic markers, including serum bilirubin levels exceeding 6mg/dl or a Mayo Risk Score greater than 7.8. (LoE 2, strong recommendation).
- •
LT should be considered in selected patients with PBC who experience severe, refractory pruritus unresponsive to medical therapy, even in the absence of hepatic decompensation or HCC. (LoE 4, strong recommendation).
- •
LT is not recommended for patients with PBC on the basis chronic fatigue in the absence of hepatic decompensation or HCC, as fatigue typically persists after transplantation. (LoE 4, strong recommendation).
In terms of survival and quality of life, LT is the most effective treatment for end-stage liver disease related to PBC (patient survival rates at 1, 3, and 5 years are 90%, 86%, and 84%, respectively).118 Nevertheless, PBC accounts for only approximately 2–6% of all LT, representing an almost 50% reduction over the past 25 years. This decline is primarily due to significant advancements in clinical management and the introduction of new therapeutic options.119,120
LT in PBC follows the general indications established for other causes of cirrhosis (decompensation and/or HCC). However, serum bilirubin level or Mayo Risk score exceeding 6mg/dl or 7.8, respectively, are considered special risk factors for PBC.121–123 Mayo risk score was the first in identifying the main risk factors for mortality in patients with PBC, and constituted an indication for LT based on age, bilirubin and albumin levels, prothrombin time and presence of edema.
It is widely accepted that patients with PBC with first decompensation and/or MELD scores>15 should be promptly considered for LT. PBC patients face a relative disadvantage in access to LT compared to those with other etiologies, because of higher waitlist mortality and lower transplantation rates. Prioritization on the LT waiting list based on the MELD score disadvantages patients with PBC, due to gender-related bias and the inconsistent recognition of refractory pruritus as a valid indication for transplantation.120,124 New models such as MELD 3.0, and GEMA-Na can amend this inequity125 but further validation of these scores in PBC patients is needed.
Should UDCA be used to prevent post-transplant PBC recurrence? Is there any benefit of an immunosuppression scheme in the prevention of PBC recurrence after LT?Recommendations:
- •
It is recommended to initiate preemptive treatment with UDCA at 10–15mg/kg/day after LT in patients with PBC, as it is associated with a reduced risk of disease recurrence and improved graft and patient survival. (LoE 3, strong recommendation).
- •
No universal immunosuppressive regimen can be specifically recommended for PBC patients undergoing LT. (LoE 3, weak recommendation).
The first report of PBC recurrence after LT was published by Neuberger et al.126 Based on a longer follow-up period, several studies have described recurrence rates ranging from 10 to 61%, with limited impact on graft and patient survival.127–129 Variability in diagnostic criteria, different follow-up times, and immunosuppressive treatments are responsible for the discrepancy in recurrence rates.129 Nevertheless, graft failure, re-transplantation, and death secondary to PBC recurrence have been reported in the literature.
Because symptoms and biochemical liver tests are non-specific and AMA persists after LT, the diagnosis must be based on liver biopsy, excluding other causes of graft dysfunction. Histological features of PBC recurrence in the graft include lymphoplasmacytic portal inflammation, lymphoid aggregates, epithelioid granulomas and evidence of bile duct injury.129,130
Several risk factors have been associated with post-liver transplant recurrence of PBC, including recipient and donor age, serum immunoglobulin M levels, HLA-DR mismatch, immunosuppression regimen (tacrolimus vs. cyclosporine), and persistent biochemical cholestasis after transplantation.120 A systematic review and meta-analysis including six retrospective studies of 3184 patients who underwent LT for PBC showed that recurrence was diagnosed in 935 (29.4%) patients, indicating that the use of tacrolimus and preventive UDCA were risk and protective factors, respectively.131 In this regard, a retrospective multicenter cohort study conducted by the Global PBC Study Group, which included 780 patients who underwent LT for PBC between 1983 and 2017, assessed disease recurrence based on histological evidence. The study found that the preventive use of UDCA and cyclosporine as immunosuppressive agents was associated with the lowest risk of recurrence and mortality.132 In another meta-analysis involving 1727 patients, Pedersen et al. demonstrated the protective effect of UDCA in preventing PBC recurrence after LT.133 However, regarding the immunosuppressive regimen, several previous studies have reported that the choice between cyclosporine and tacrolimus does not influence recurrence rates.134,135
Currently, there are no clear guideline recommendations regarding immunosuppressive regimens for PBC recurrence prevention. When selecting between cyclosporine and tacrolimus, it is essential to consider their potential side effects, including risks of acute and chronic rejection, infection, and renal dysfunction. Therefore, treatment decisions should be made on an individualized basis, weighing the benefits and risks of each regimen.136
How is PBC/AIH variant diagnosed?Recommendation:
- •
The Paris criteria remain the best-validated and guideline-endorsed system for diagnosing PBC/AIH variant. However, Zhang score may offer improved sensitivity in milder presentations (LoE 4, strong recommendation).
- •
A Zhang scoring system>21 should prompt the diagnosis of PBC-AIH variant (LoE 4, strong recommendation).
PBC and AIH are distinct clinical entities characterized by specific biochemical, histological and serological profiles.137,138 However, there is a group of patients with features of both AIH and PBC at diagnosis139–141 or after several years of an initial diagnosis of PBC142–144 or AIH.142
The Paris criteria is the most commonly used tool for diagnosing variant syndrome (VS), formerly known as PBC/AIH overlap syndrome.145 This requires the presence of at least two of three key criteria for the diagnosis of PBC and AIH. Thus, for PBC, the criteria are as follows: (1) ALP>2× ULN or GGT>5× ULN; (2) positive AMA; and (3) liver biopsy specimen showing florid bile duct lesions. For AIH, the criteria are as follows: (1) ALT levels>5× ULN; (2) serum immunoglobulin G (IgG) levels>2× ULN or positive anti-smooth muscle antibodies (ASMAs); and (3) liver biopsy showing moderate or severe interface hepatitis. The 2009 EASL guidelines for the management of cholestatic liver diseases endorsed the Paris criteria while emphasizing that histologic evidence of moderate to severe interface hepatitis is essential for the diagnosis of VS.146 The reported sensitivity and specificity of the Paris criteria for VS are 92% and 97%, respectively.145 The reported sensitivity and specificity of the Paris criteria for VS are 92% and 97%, respectively.147,148 Furthermore, during the natural progression of PBC, all untreated patients develop interface hepatitis within four years, and a considerable proportion demonstrate positive ANA and/or ASMA at diagnosis.149 These findings indicate that meeting the diagnostic standards for PBC combined with significant interface hepatitis and presence of ANA and/or ASMA does not suffice to define VS.
The International AIH Group (IAIHG) scoring system for AIH is another widely used diagnostic criterion for the diagnosis of VS, although it was not intended for such use and has not proven to be an efficient tool for this purpose.139 The diagnostic criteria for AIH were initially introduced by the IAIHG in 1993 and later revised in 1999.43,150 The revised IAIHG scoring system was able to identify 19% of AIH variant when applied to 137 patients diagnosed with PBC.151,152 The IAIHG suggests that patients with autoimmune liver disease should be categorized according to their predominant features and that patients with variant features should not be considered distinct diagnostic entities.
Consequently, there is a pressing need for the development of novel diagnostic criteria capable of detecting both typical and mild VS with high sensitivity and specificity. This is of paramount importance, as research indicates that patients with VS may experience poorer clinical outcomes compared to those with PBC alone, including an earlier onset of PH and an increased likelihood of requiring LT.149 To overcome these limitations a new scoring system for the identification of VS has been recently developed by modifying the IAIHG revised criteria.151 The “Zhang criteria” score integrates the histological features of AIH and PBC, alongside modifications in biochemical and immunological characteristics. The optimal diagnostic performance for VS was observed in patients with a score of ≥21, yielding a sensitivity of 98.5%, specificity of 92.8%, positive predictive value (PPV) of 81.0%, and negative predictive value (NPV) of 99.5%. All individuals diagnosed with VS had an aggregate score exceeding 19, whereas the majority of patients with PBC or AIH scored below 19, establishing this as an effective discriminatory threshold against VS. In assessing the sensitivity of the Paris criteria for diagnosing VS within this cohort, the sensitivity was 58.46%, specificity was 99.52%, PPV was 97.44%, and NPV was 88.41%. Compared to the Paris criteria, the Zhang diagnostic criteria, with a cut-off score of 21, demonstrated superior sensitivity and NPV but lower specificity and PPV. This classification method appears to accurately identify patients with VS and may surpass existing scoring systems, including the revised and simplified AIH scores and the Paris criteria, in detecting VS and its milder forms. However, the Zhang criteria were developed in a study by Zhang et al. that included a limited sample size from a single center, necessitating further studies to assess its validity. Despite the enhanced precision of Zhang's criteria, variant forms remain difficult to classify. A retrospective study evaluating patients diagnosed with VS revealed that only 24% met the Paris criteria and only 70% met Zhang's criteria, indicating that current classification tools are suboptimal. Furthermore, a significant finding of the study was that patients who did not meet the VS criteria and were not treated with combined immunosuppressive therapy and UDCA, but exhibited analytical markers of inflammation (defined as ALT or AST>2.5× ULN and elevated IgG), had a poorer prognosis than those classified as VS who received treatment, even if they did not meet the Paris or Zhang criteria.153
Autoantibodies, particularly anti-dsDNA, have been proposed as useful non-invasive markers for identifying patients with VS,138 given their higher prevalence in VS compared to PBC alone. Finally, a recent study developed two artificial intelligence-based predictive models using serological data from 201 biopsy-proven cases of autoimmune liver disease.137 Showing the potential utility of IgG, IgM, anti-Sp100, anti-Ro-52, anti-SSA, ANA, partial thromboplastin time and α-fetoprotein as additional discriminative variables for diagnosing VS. However, these results need further evaluation and external validation.
What are the most frequently associated symptoms in PBC? How should they be assessed? What therapeutic alternatives are available?
Recommendations:
- •
In patients with PBC, the presence and severity of pruritus and fatigue should be assessed at every clinical visit (LoE 4, strong recommendation).
- •
Bezafibrate is the recommended first line treatment of moderate-to-severe pruritus (LoE 2, strong recommendation).
- •
Ileal bile acid transporters inhibitors should be considered as an alternative to bezafibrate, particularly in patients with well-controlled disease (LoE 2, strong recommendation).
- •
In the management of fatigue, aerobic and resistance physical exercise is recommended as a non-pharmacological intervention (LoE 3, strong recommendation).
Pruritus is a highly prevalent symptom in patients with PBC, affecting up to 70–80% of them at some point during the disease course. Nevertheless, its expression varies greatly, as some patients experience mild and intermittent itching, while others suffer from severe, chronic pruritus that significantly impairs quality of life. Importantly, pruritus may occur even in early-stage or biochemically well-controlled PBC and does not necessarily correlate with liver enzyme levels or histological activity.
The pathogenesis of cholestatic pruritus in PBC is not completely understood. A key mechanism involves the accumulation of BA and lysophosphatidic acid (LPA),154 produced by the enzyme autotaxin. These compounds are believed to stimulate pruritogenic receptors on cutaneous and sensory neurons. The G-protein-coupled receptor MRGPRX4 has emerged as a likely candidate mediating bile acid-induced itch.155 Additionally, elevated endogenous opioids and an imbalance in μ- and κ-opioid receptor activity are thought to modulate central itch perception. Other contributors may include bilirubin, histamine-independent pathways, and neural sensitization, particularly in chronic cases.156 However, the relative roles of these mediators remain debated, and not all patients with high levels of BA or autotaxin experience pruritus, suggesting a complex interplay of mechanisms.
As the prevalence of pruritus is very high, an adequate evaluation of pruritus and its impact on quality of life is of critical importance. The most used tools include the Numerical Rating Scale (NRS) and the Visual Analog Scale (VAS), where patients rate their itch intensity on a scale from 0 (no itch) to 10 (worst imaginable itch). Multidimensional tools such as the 5-D Itch Scale (assessing duration, degree, direction, disability, and distribution)157 and disease-specific instruments like the PBC-40 questionnaire provide a broader evaluation, incorporating the impact of pruritus on quality of life, sleep, and emotional well-being.158
Once the severity of pruritus is adequately evaluated, the next step is to decide the best treatment alternative for the patient (Fig. 3). Classically, the standard initial therapy was cholestyramine, a bile acid sequestrant that interrupts enterohepatic recirculation of BA. It is typically administered at 4g once or twice daily (up to 16g/day), taken apart from other medications to avoid absorption interference. Although often effective in mild cases, its texture and gastrointestinal side effects usually limit long-term use. Consequently, cholestyramine is increasingly being replaced by bezafibrate, the efficacy of which was demonstrated in the FITCH trial.159 The study conducted in patients with severe pruritus showed that bezafibrate significantly reduced pruritus compared to placebo and also improved other quality-of-life parameters, such as sleep. Based on these results, as well as the accumulating evidence from real-world clinical practice,160 bezafibrate is considered first-line pharmacologic treatment. Another therapeutic option is rifampicin,161 which acts by activating PXR, thereby inducing hepatic enzymes that promote the clearance of pruritogens such as autotaxin. The typical dosage ranges from 150 to 300mg once or twice daily, with the onset of action occurring within days to weeks. Close monitoring of liver function is essential due to the potential risk of hepatotoxicity. Naltrexone,162 a μ-opioid receptor antagonist, at 12.5–25mg/day and sertraline163 (selective serotonin reuptake inhibitor) at doses of 75–100mg/day, have demonstrated moderate antipruritic effects.

Recently, ileal bile acid transporters (IBATs) inhibitors have shown relevant results in the setting of PBC-related pruritus. The efficacy of linerixibat, an IBAT which acts by blocking bile acid reabsorption in the terminal ileum, was evaluated in the GLISTEN phase 3 clinical trial. Two hundred thirty-eight patients with moderate to severe pruritus were randomized to linerixibat 40mg/day or placebo. After 24 weeks of treatment, patients in linerixibat presented a statistically significant reduction in the worst-itch NRS as compared to those on placebo (−2.86 vs. −2.15, p=0.0013). A larger proportion of linerixibat-treated patients reported their pruritus had very much improved at week 24 (54.6% vs. 37.3%). Gastrointestinal side effects were more frequently observed in patients treated with linerixibat but treatment discontinuations (mainly due to diarrhea) were very low (7% vs. <1% in the placebo group).164
MARS (Molecular Adsorbents Recirculating System) and plasma exchange have been used as rescue therapies for intractable pruritus in PBC by removing circulating pruritogens. Although their effects are temporary, they can provide significant symptomatic relief in patients unresponsive to conventional pharmacological treatments. LT may be also considered in the setting of severe refractory pruritus.8
Finally, as mentioned before, the effect on pruritus of the recently approved PPAR agonists, seladelpar and elafibranor, was evaluated as secondary endpoint in the RESPONSE and ELATIVE trials, respectively. Treatment with seladelpar was an associated with a significant decrease in the weekly mean NRS score at 6 months of treatment in patients with moderate to severe pruritus at baseline. Elafibranor also decreased NRS score at 6 months, but the difference did not reach statistical significance. More data from clinical trials and real-life cohorts are needed to confirm these results. In the meantime, seladelpar could serve as an alternative drug for patients with intolerance or contraindications to bezafibrate.
FatigueFatigue is a prevalent and disabling symptom in PBC, affecting up to 78% of patients regardless of disease stage, and is a major contributor to impaired quality of life. Notably, fatigue does not correlate with histological disease activity, suggesting the involvement of mechanisms beyond hepatic damage. The causes of fatigue in the setting of PBC are not known. Current evidence supports multifactorial pathogenesis, with two principal non-mutually exclusive hypotheses proposed. The first is the “peripheral fatigue hypothesis”, which suggests that mitochondrial dysfunction impairs aerobic muscle metabolism, leading to an early shift toward anaerobic energy production during exertion and delayed post-exercise recovery.165–167 The second is the “central fatigue hypothesis”, which attributes fatigue to central nervous system dysfunction characterized by cognitive impairment, excessive daytime sleepiness, and altered functional connectivity in key brain regions. This central dysfunction is thought to result from chronic neuroinflammation and neurosteroid imbalances.168,169 Supporting this view, neurophysiological studies have demonstrated impaired central motor activation in PBC patients, a phenomenon that persists even after LT, thereby reinforcing the concept of a central, liver-independent mechanism. Clinically, the expression of fatigue is heterogeneous, with some patients experiencing predominantly peripheral manifestations and others exhibiting central cognitive deficits.
Assessment of fatigue depends on patient-reported outcome measures, as no objective biomarker or diagnostic test currently exists for this symptom. Among the validated tools, the PBC-40,158 which includes a dedicated fatigue domain, has been extensively used in clinical trials and observational studies. Objective assessments, including handgrip strength and exercise tolerance tests like the shuttle walk test, may provide additional insight into fatigue-related performance deficits. Furthermore, it is important to evaluate excessive daytime sleepiness separately, as it may coexist with or mimic fatigue but may require different therapeutic approaches.
In terms of treatment, managing fatigue in PBC is challenging, as no approved or consistently effective medication exists to date.170–172 However, post hoc analyses of the RESPONSE and ELATIVE trials have demonstrated an improvement in the fatigue domain of the PBC-40 in patients treated with seladelpar and elafibranor, with the improvement being statistically significant for elafibranor. Furthermore, setanaxib, a dual inhibitor of NOX1 and NOX4 currently under evaluation in clinical trials, has shown potential in ameliorating fatigue in PBC.173 However, further studies are required to confirm its long-term efficacy and safety. Considering this, the non-pharmacological approach is the cornerstone. A fundamental component of this approach is patient education and validation, as acknowledging the legitimacy and burden of fatigue can itself be helpful and somehow therapeutic. Patients frequently struggle with the invisible and misunderstood nature of their symptoms and then, a supportive and empathetic clinician-patient relationship is essential for improving trust and facilitating effective interventions. In parallel, energy conservation techniques are important to daily symptom management. Patients should be encouraged to implement structured pacing strategies, such as prioritizing tasks during peak energy periods, scheduling rest intervals, and using adaptive tools for routine activities. Occupational therapy can assist in reinforcing these practices. Among all non-pharmacological interventions, exercise therapy has emerged as one of the most effective approaches for alleviating fatigue in PBC. Despite initial apprehension among patients that physical activity might worsen exhaustion, evidence suggests that inactivity leads to progressive deconditioning and increased muscle inefficiency. A pivotal proof-of-concept trial involving 31 PBC patients with moderate-to-severe fatigue demonstrated that a 12-week home-based program, consisting of individualized aerobic and resistance exercises, resulted in a clinically significant reduction in fatigue in 84% of participants, defined as a ≥5-point improvement in the PBC-40 fatigue domain, with a median reduction in fatigue score of approximately 10.5 points.174 Moreover, participants experienced measurable gains in exercise capacity, including improved walk distance and peak oxygen consumption, alongside subjective improvements in cognitive clarity and reduced daytime somnolence.
What PBC treatment should be recommended during pregnancy?Recommendations:
- •
UDCA is the treatment of choice for PBC in pregnant women. (LoE 4, strong recommendation).
- •
UDCA should not be discontinued or modified during pregnancy. It is safe for mothers, fetuses, and deliveries. (LoE 4, strong recommendation).
Clinical experience with the management of PBC during pregnancy remains limited, and available evidence regarding treatment strategies and follow-up is scarce. UDCA is currently the recommended first-line therapy for PBC in pregnant patients.8 It is considered safe, with no reported severe adverse effects on maternal health, fetal development, or delivery outcomes.175–178 Notably, UDCA also constitutes the treatment of choice for intrahepatic cholestasis of pregnancy (ICP).179 International guidelines support the continuation of UDCA therapy throughout pregnancy in patients with PBC.8
Rifampicin, which has not been associated with teratogenic or other fetal adverse effects, is a suitable therapeutic alternative for patients with refractory pruritus who do not respond adequately to cholestyramine.180
There is an absence of clinical data on the safety of bezafibrate in pregnant women. Based on preclinical findings suggesting potential teratogenicity,181 bezafibrate is currently contraindicated during pregnancy according to the prescribing information.182 Nevertheless, isolated case reports have described its off-label use in the third trimester of pregnancy for severe hypertriglyceridemia183,184 and, in rare instances, for pruritus associated with ICP,185 although this information should be interpreted with caution.
There is no information regarding the safety or elafibranor1 and seladelpar2 during pregnancy. At the time of publication of this document, the Spanish Agency of Medicines (AEMPS) states in the Summary of Product Characteristics (SmPC) for elafibranor that it is contraindicated during pregnancy and must be discontinued if a woman becomes pregnant. Although there is no evidence of teratogenic effects in animal studies, the SmPC for seladelpar indicates that it is advisable to avoid its use during pregnancy as a precautionary measure.
Does the presence of PBC affect pregnancy outcomes such as fetal death, malformations, preterm birth, or maternal mortality?Recommendation:
- •
It is suggested that increased clinical monitoring during pregnancy and the postpartum period is necessary because of a higher risk of biochemical deterioration and pruritus. (LoE 3, strong recommendation).
- •
Patients with PBC-related cirrhosis have an increased risk of developing complications. Prophylactic and therapeutic management of cirrhosis and its complications, like that of other patients with cirrhosis of any etiology, is recommended. (LoE 5, strong recommendation).
Pregnancy is generally well-tolerated in patients with PBC. However, it is associated with increased fetal morbidity. A recent meta-analysis demonstrated that women with PBC are at a higher risk of preterm delivery.186–188 Moreover, pregnancy may be accompanied by exacerbation of pruritus or development of de novo pruritus during gestation.187,189 A significant proportion of patients also experience biochemical flares, characterized by worsening cholestasis during the postpartum period,178 underscoring the importance of close clinical monitoring in the weeks following delivery.178 Importantly, no cases of maternal mortality have been reported during pregnancy or postpartum period in women with PBC.
In contrast, pregnancy in patients with PBC complicated by cirrhosis entails a substantially elevated risk of adverse outcomes for both the mother and the fetus. In particular, the presence of PH increases the likelihood of variceal hemorrhage. For such high-risk cases, multidisciplinary monitoring and management by hepatology and obstetrics specialists is strongly recommended.8
Primary sclerosing cholangitis in adultsHow is PSC diagnosed?Recommendation:
- •
In adult patients presenting with elevated serum markers of cholestasis, a diagnosis of large-duct PSC should be made in the presence of typical findings of sclerosing cholangitis on high-quality cholangiography and after the exclusion of secondary causes. The preferred diagnostic test is MRCP. (LoE 2, strong recommendation).
- •
A diagnosis of small-duct PSC should be considered in patients with elevated serum markers of cholestasis of unknown cause, normal high-quality cholangiography, and compatible histology of PSC, particularly in those with concomitant IBD. (LoE 3, strong recommendation).
- •
Autoantibodies should not be used to diagnose or stratify patients with PSC. (LoE 4, strong recommendation).
PSC is a chronic immune-mediated liver disease characterized by inflammation, fibrosis, and destruction of the intrahepatic and/or extrahepatic bile ducts, leading to cholestasis, bile duct strictures, and liver fibrosis, with potential progression to cirrhosis, PT, and liver decompensation.3,190–193 There is a variant known as small-duct PSC, characterized by cholestasis along with typical histological findings of PSC, but with a normal appearance of the bile ducts on cholangiography. Furthermore, PSC can coexist with AIH, currently defined as PSC/AIH variant.3,190–193
PSC should be considered in any patient presenting with cholestasis, particularly if they have IBD, as up to 80% of PSC patients also have IBD in western countries.3,190–193 MRCP is preferred for diagnosis because of its non-invasive nature and high diagnostic accuracy, including sensitivity and specificity in identifying multifocal strictures and dilations in the bile ducts (“beaded appearance”), which are characteristic of PSC.3,190–194 A meta-analysis found that the pooled sensitivity and specificity of MRCP for diagnosing PSC were 86% and 94%, respectively.195 However, MRCP findings in PSC can vary significantly, likely reflecting the different stages of the disease. Therefore, repeat high-quality MRI or MRCP is recommended for patients with inconclusive cholangiographic findings or ongoing diagnostic uncertainty. The preferred protocol includes 3D MRI/MRCP with axial T1- and T2-weighted sequences and contrast enhancement.194 It is important to highlight that normal ultrasound or CT findings do not rule out PSC. Plus, it is noteworthy that MRCP also avoids the risks of invasive procedures, such as infections and pancreatitis, which are associated with endoscopic retrograde cholangiopancreatography (ERCP).196 ERCP should only be used when MRCP does not provide sufficient diagnostic information or when tissue sampling and/or therapeutic intervention are necessary. The differential diagnosis of PSC includes several conditions that cause biliary strictures, such as bacterial cholangitis, transarterial chemoembolization, prior biliary surgery, AIDS-related cholangiopathy, sclerosing cholangitis in critically ill patients, COVID-19-related cholangiopathy, IgG4 sclerosing cholangitis, and malignant conditions such as cholangiocarcinoma (CCA) or checkpoint inhibitor-induced liver injury.3,190–193 Additionally, a small subset of patients may exhibit typical sclerosing cholangitis findings on cholangiography despite normal serum ALP and GGT levels, and such patients require close clinical follow-up.190
Is liver biopsy necessary in patients with suspected PSC and normal MRCP findings?Recommendations:
- •
In adults suspected of having PSC, liver biopsy should be performed if high-quality MRCP is normal to confirm or exclude small-duct PSC. (LoE 4, strong recommendation).
- •
Liver biopsy should be considered in patients with PSC and coexisting features of AIH (LoE 4, strong recommendation).
In approximately 10% of cases, PSC affects only the peripheral ducts and is not visible on MRCP or ERCP; this condition is known as small-duct PSC. When suspected, a liver biopsy should be performed, especially in the context of concomitant IBD. Small duct PSC is characterized by periductal fibrosis and fibro-obliterative duct lesions on histological examination. Additional compatible features include bile duct loss, ductular reaction (also known as ductular proliferation), biliary pattern of interface activity, and chronic cholestatic changes in the periportal hepatocytes.197,198 Concentric “onion skin” periductal fibrosis, although less common, can be seen in PSC as well as in other obstructive cholangiopathies. Several fibrosing small duct cholangiopathies can mimic the histopathological findings of small duct PSC, including a genetic ATP-binding cassette B4 (ABCB4) deficiency. Therefore, thorough evaluation is essential before diagnosing small-duct PSC, especially in the absence of IBD.
Is it necessary to determine IgG4 levels in patients with suspected PSC?Recommendations:
- •
The determination of serum IgG4 levels is suggested in every adult patient with large-duct sclerosing cholangitis at the time of diagnosis (LoE 3, weak recommendation).
- •
It is recommended that the diagnosis of IgG4-SC be conducted in accordance with the guidelines established by the international consensus on IgG4-related disorders. (LoE 2, strong recommendation).
Serum IgG4 levels are elevated in up to 15% of patients with PSC, although their clinical significance remains unclear. Although rare, markedly elevated IgG4 levels (>5.6g/L) are suggestive of IgG4-related sclerosing cholangitis (ISC).199 It is important to remember that a diagnosis of PSC should only be established after excluding the potential causes of secondary sclerosing cholangitis. ISC, like PSC, is associated with biliary strictures, elevated serum markers of cholestasis, and increased serum IgG4 levels, along with similar clinical signs and symptoms, making differentiation challenging.199,200 However, ISC is more commonly found in elderly men, particularly those with long-term exposure to potentially harmful chemicals.201 The diagnosis of ISC can be established using HISORt criteria, which are based on histological, imaging, and serological (IgG4) findings, involvement of other organs (such as autoimmune pancreatitis and sialadenitis), and response to corticosteroid treatment.199,200
When should a PSC/AIH variant be suspected?Recommendations:
- •
A PSC/AIH variant should be suspected in patients with cholangiographic features of PSC in combination with findings suggestive of AIH including markedly elevated transaminases, high IgG levels, and positive autoantibodies. (LoE 4, strong recommendation).
- •
A liver biopsy should be considered in people with PSC and coexisting features of AIH. (LoE 4, strong recommendation).
Liver biopsy should be considered for patients with PSC who exhibit features suggestive of AIH, such as markedly elevated transaminases (>5× ULN), high IgG levels (>1.5–2× ULN), and positive autoantibodies compatible with AIH (ANA, SMA, and/or liver/kidney microsomal antibodies [LKM). However, hypergammaglobulinemia, autoantibodies, and elevated serum transaminase levels are frequently observed in both AIH and PSC. Indeed, elevated transaminase levels are frequently noted in PSC and do not necessarily indicate an AIH variant unless they are predominant. Furthermore, the presence of serum autoantibodies in patients with PSC is highly variable and likely indicative of underlying immune dysregulation. Additionally, mild interface hepatitis may be observed in liver biopsies of patients with PSC. However, the presence of moderate-to-severe lymphoplasmacytic interface hepatitis in a patient with known PSC suggests the coexistence of AIH. The prevalence of the AIH variant in adults diagnosed with PSC ranges from 1.4% to 17%. Conversely, a small percentage of patients with AIH may show PSC features. These patients typically present with cholangiographic features of PSC alongside AIH features, including younger age, higher transaminase levels, elevated IgG, positive autoantibodies, and mixed histopathological changes, such as interface hepatitis and the typical biliary pathology of PSC.202
What methods are employed to assess the severity and prognosis of PSC? What is the role of prognostic score systems and surrogate markers as key tools for monitoring disease progression?
Recommendations:
- •
Risk assessment should be performed at diagnosis and during the follow-up. (LoE 3, strong recommendation).
- •
Standard biochemical parameters, such as serum bilirubin, albumin, ALP, ALT, platelets, and prothrombin time, should be determined at baseline and during follow-up. (LoE 3, strong recommendation).
- •
Liver ultrasound and/or abdominal MRI/MRCP should be performed annually. (LoE 3, strong recommendation).
- •
Liver elastography and/or serum fibrosis tests should be performed at least every 2–3 years. (LoE 3, strong recommendation).
Given the heterogeneity of PSC and the typically prolonged interval between diagnosis and clinical endpoints, such as progression to cirrhosis, liver-related complications, and CCA, the development of prognostic models is essential.3 In 2016, a publication from the International PSC Study Group highlighted the existing limitations and emphasized the necessity for improved surrogate endpoints, as the available data did not attain level 2 validation for any biomarkers (i.e., validation as a surrogate endpoint).203 The challenges associated with percutaneous liver biopsy in PSC, including sampling variability and the limitations of histological scoring systems, particularly the lack of validation for PSC-specific criteria, have contributed to the declining use of histological evaluation.204 Consequently, recent efforts have focused on the development of non-invasive prognostic tools for PSC, including biomarkers, non-invasive fibrosis tests, and biliary imaging studies.3,205
Serum markersALP is the most extensively studied biomarker, both independently and in combination with the assessment of liver stiffness using elastography or MRCP.3 ALP reduction has emerged as a critical factor associated with improved TFS in PSC.206 In a prospective cohort study of 215 patients with PSC, 59.5% and 56.7% met the ALP reduction criteria, defined as normalization, a 50% decrease, or a reduction to below 1.5× ULN within 6 months and a reduction to below 1.5× ULN within 12 months, respectively. Meeting the endpoints was associated with prolonged TFS in all patients, regardless of the presence of dominant strictures. Furthermore, studies examining ALP normalization in PSC have underscored its prognostic relevance.207,208 Patients achieving ALP normalization within 12 months exhibit extended survival and a reduced risk of requiring LT, with persistent normalization further improving these outcomes.207,208 Moreover, another study identified an optimal threshold for ALP levels, demonstrating its discriminatory ability between patients with good and poor prognoses.209
The progressive nature of PSC is characterized by spontaneous bilirubin fluctuations that may be triggered by events such as acute bacterial cholangitis, biliary stones, or the development of high-grade strictures.3 This dynamic course presents challenges for accurately predicting prognosis using conventional models, such as the Child–Pugh and MELD scores.3,210 Consequently, the complex relationship between total bilirubin levels and disease progression highlights the challenges in predicting clinical outcomes in patients with PSC.3,210 Although elevated total bilirubin levels have been associated with reduced survival, existing studies demonstrating this correlation have predominantly involved patients with advanced disease, thereby limiting its applicability to early-stage PSC.211,212
The Enhanced Liver Fibrosis (ELF) score and ELF test, originating from algorithms involving tissue inhibitor of metalloproteinase I, hyaluronic acid, and propeptide of type III procollagen, have been studied in the context of PSC. In a cohort of patients with PSC, the ELF score demonstrated independence from the Mayo Risk Score and exhibited a c-statistic of 0.82 for TFS, with a score of ≥10.6 linked to lower TFS. Conversely, the ELF test outperformed the ELF score in identifying low-risk groups for clinical endpoints.213 In a clinical trial comparing simtuzumab and placebo in PSC patients, an ELF test≥9.8 was associated with PSC-related progression events.214 In relation to CCA, researchers have identified a notable increase in the ELF score among affected patients, irrespective of underlying chronic liver disease, disease stage, or age. This finding suggests that the ELF score may serve as a potential tool for risk stratification of CCA in PSC patients.215
Non-invasive prognostic modelsThe Mayo Risk Score incorporates age, bilirubin, AST, variceal bleeding, and albumin into its predictive formula.
This biopsy-free model, independently validated, demonstrated a strong correlation with actual survival over a 4-year period.216 Since then, numerous prognostic models have been developed, and their accuracy has been compared with that of the Mayo Risk Score.
Based on data from 1001 patients, the UK-PSC risk score, which incorporates variables such as age, bilirubin, ALP, albumin, platelet count, extrahepatic biliary disease, and variceal hemorrhage, demonstrated superior performance compared to the Mayo and APRI scores after external validation. Serum ALP ≥2.4× ULN at 1 year and the presence of extrahepatic biliary disease were identified as predictors of adverse 10-year outcomes.217
The PSC risk estimate tool (PREsTo) model, developed using gradient boosting and validated in both North American and international multicenter cohorts, has emerged as an effective predictor of hepatic decompensation in PSC. The PREsTo model outperforms existing surrogate markers, maintaining high accuracy across a range of clinical scenarios, including individuals with bilirubin<2.0mg/dl and when reapplied later in the disease course, and offers superior predictive performance for hepatic decompensation compared to other non-invasive prognostic tools.204
The Amsterdam–Oxford Model, developed and externally validated in a large, population-based cohort, provides a broadly applicable prognostic tool for predicting TFS in PSC.205,206 Utilizing easily accessible variables (age, albumin, bilirubin, AST, ALP, platelets, history of ulcerative colitis, previous history of infectious cholangitis and/or biliary surgery) this model demonstrated satisfactory discrimination and calibration, showing robustness in estimating PSC-related death and/or LT. Additionally, the model demonstrates consistent predictive performance when applied to follow-up biochemical parameters.205,206
In conclusion, these advancements reflect the ongoing evolution of prognostic modeling of PSC, with each model offering distinct insights and clinical applications (Table 3).
Prognostic models with demonstrated predictive capacity in patients diagnosed with PSC.
| Prognostic Model | Variables | Endpoint |
|---|---|---|
| Mayo Risk Score | Age, Bilirubin, AST, Variceal Bleeding, Albumin | Survival up to 4 years |
| UK-PSC Risk Scores | Age, Bilirubin, Alkaline Phosphatase, Albumin, Platelets, Extrahepatic Biliary Disease, Variceal Hemorrhage | 2-Year and 10-year outcomes |
| PREsTo (PSC Risk Estimate Tool) | Bilirubin, Albumin, SAP x ULN, Platelets, AST, Hemoglobin, Sodium, Age, Years Since PSC Diagnosis | Hepatic Decompensation in PSC |
| Amsterdam-Oxford Model | PSC Subtype, Age at PSC Diagnosis, Albumin, Platelets, AST, Alkaline Phosphatase, Bilirubin | Transplant-free Survival in PSC |
MRI is the imaging modality of choice in patients with PSC, given its ability to noninvasively evaluate the biliary tree and potentially assess disease progression. In 2014, a French study introduced the ANALI score, derived from standardized MRI interpretations. This score demonstrated a strong association between radiologic progression and adverse clinical outcomes.218 Two variants of the ANALI score, with and without gadolinium, were subsequently developed and externally validated. Both versions showed prognostic value, with higher scores significantly associated with an increased risk of liver-related death, LT, or hepatic decompensation. Specifically, the non-gadolinium variant conferred a 25-fold increased risk, and the gadolinium-based score a 13-fold increase, compared to lower scores.219
Despite these promising findings, clinical applicability of the ANALI score remains limited. In a study of 98 PSC patients, expert radiologists reported only modest inter-reader agreement. This was particularly evident for the non-gadolinium version, which showed poor-to-moderate reproducibility (ICC=0.56), thus challenging its reliability in routine practice. These findings highlight the need for broader multicenter validation before widespread adoption.220
To address these limitations, novel MRI-based tools have emerged, leveraging advanced image analysis and artificial intelligence. One such tool is the quantitative MRCP (qMRCP) score, which has shown superior performance over the traditional ANALI score in identifying patients at higher risk for hepatobiliary complications.221 Additionally, MRCP+, a three-dimensional analysis platform, has demonstrated enhanced accuracy in distinguishing normal from abnormal bile ducts. It provides valuable insights into disease severity, risk stratification, and progression dynamics.222 Finally, a newer cholangiographic scoring system, known as DiStrict, has been specifically designed for large-duct PSC. Based on MRCP findings, this score has shown strong prognostic value, is highly reproducible, and is emerging as a simple yet powerful tool for predicting outcomes in this PSC phenotype.223
Liver stiffness measurementLSM is a widely available and validated non-invasive tool for assessing liver fibrosis across a broad spectrum of chronic liver diseases. Although its ability to precisely stage fibrosis may vary depending on the underlying etiology, LSM has demonstrated consistent predictive value for clinical outcomes in various clinical settings.
In the context of PSC, several studies have evaluated the role of LSM as both a diagnostic and prognostic tool. A prospective study of 73 PSC patients followed over four years showed that LSM correlated independently with histological fibrosis stage and outperformed conventional scores such as APRI, FIB-4, and the Mayo risk score in identifying significant and severe fibrosis. Furthermore, LSM displayed high reproducibility across operators and anatomical measurement sites. Notably, both baseline LSM values and the rate of stiffness progression were strongly associated with clinical outcomes in patients treated with UDCA.224
Complementary findings emerged from a study of 30 PSC patients undergoing LT evaluation, in which TE values correlated significantly with the Laennec fibrosis stage, collagen content, and septal thickness in explanted livers. TE also showed strong associations with biochemical markers of liver injury (e.g., AST, bilirubin, FIB-4, and APRI). In multivariate analysis, histological fibrosis, assessed via Laennec staging or collagen quantification, was the primary determinant of LSM values. Importantly, TE proved particularly effective in identifying cirrhosis, with a cutoff of 13.7kPa showing excellent diagnostic accuracy.225
Further validation comes from the simtuzumab trial,214 in which the optimal LSM cutoffs for advanced fibrosis (≥9.6kPa) and cirrhosis (≥14.4kPa), previously reported by Corpechot et al.,224 were confirmed to have good and excellent diagnostic performance, respectively. Additionally, MR elastography (MRE) has been explored in a cohort of 20 patients with biopsy-confirmed PSC, showing excellent accuracy for fibrosis staging. However, these promising results require confirmation in larger, independent cohorts before clinical integration.226
Taken together, this body of evidence supports the use of TE as a robust, non-invasive tool for staging liver fibrosis and estimating prognosis in PSC. Nonetheless, it is important to be aware of that obstructive cholestasis due to untreated dominant strictures of the common bile duct or hepatic ducts can lead to falsely elevated LSM values.227
Novel prognostic biomarkersMacrophage activation markers such as soluble CD163 (sCD163) and soluble mannose receptor (sMR) have shown a strong correlation with disease severity. Elevated levels of these markers are associated with shorter TFS, especially in patients with more advanced disease.228 In addition, proteomic analysis of bile samples from Norwegian and Finnish cohorts identified 14 proteins differentially expressed between PSC patients and controls, with IL-8 and S100A12 notably linked to PSC diagnosis and cholangiographic severity. Serum IL-8 levels demonstrated robust discriminatory power for TFS in both derivation and validation cohorts and remained independently associated with prognosis after adjusting for age and disease duration.229 However, the ELF and Mayo risk scores showed superior predictive accuracy for TFS in the same study.229 Autotaxin has also emerged as a promising biomarker, validated in two independent Norwegian cohorts of large-duct PSC patients, where increased autotaxin activity independently predicted reduced TFS.230
Beyond protein-based markers, PSC has also been associated with alterations in plasma bile acid (BA) profiles, which may be influenced by UDCA treatment and coexisting IBD. Elevated concentrations and conjugated fractions of several BA species were linked to a higher risk of hepatic decompensation, while a higher glycine-to-taurine conjugation ratio showed a protective effect.231
How can a dominant or high-grade stricture be defined and treated in patients with PSC?Recommendations:
- •
The luminal narrowing of >75% of the bile duct should be classified as high-grade stenosis (HGS) (LoE 3, weak recommendation).
- •
Clinically relevant stenosis can be defined as stenosis accompanied by symptoms such as jaundice and cholangitis. (LoE 3, weak recommendation).
- •
In patients with HGS cytology and brushing should be performed during ERCP. To improve diagnostic accuracy, advanced techniques such as FISH, digital image analysis, flow cytometry, or next-generation sequencing may be considered, depending on individual case characteristics (LoE 2, strong recommendation).
- •
It is recommended to use balloon dilation as the first-line intervention for managing HGS in patients with PSC. Repeated balloon dilation provides a viable long-term solution with acceptable complications and positive TFS rates. (LoE 2, strong recommendation).
- •
Routine stenting after balloon dilation is discouraged owing to a lack of clear benefits and a higher risk of complications. (LoE 2, strong recommendation).
More than 50% of patients will develop focal HGS, formerly known as dominant stenosis (DS), during the follow-up. Defined specifically in the context of changes observed during ERCP, the term DS was arbitrarily defined as an intraluminal diameter<1.5mm in the common bile duct and <1mm in hepatic ducts within 2cm of the bifurcation.232,233 However, studies have revealed variability in these estimates, emphasizing the limitations of the DS definition. Notably, the definition of DS in ERCP is often not applicable in MRCP, particularly within the extrahepatic ducts.232 This limitation arises from the insufficient spatial resolution of MRCP, and the absence of the hydrostatic pressure applied during ERCP procedures.194,234 Additionally, MRCP may detect additional strictures peripheral to a non-traversed or poorly filled HGS on ERCP.194,234 Furthermore, bile duct wall thickening, a valuable feature in stricture assessment, is easily visualized with MRCP but is not possible on ERCP. Nonetheless, despite the association of severe strictures with morbidity, clinical perception and management vary widely among clinicians treating these lesions.194 Consequently, the use of the term DS in the evaluation of MRCP is not recommended.
A consensus among experts197,234 has been established to define criteria suitable for MRCP application. The EASL guidelines already address the concept of dominant stenosis in ERCP and introduces the distinctions of low or HGS (≥75% luminal narrowing) or clinically relevant stenosis (when associated with cholangitis or cholestasis).197,234 Nevertheless, ERCP plays a crucial role in the management of HGS in patients with PSC.3,235,236 Chronic biliary obstruction resulting from HGS holds the potential to accelerate hepatic fibrosis and cirrhosis, and reversal of this cholestatic injury may be achieved by relieving the biliary obstruction.237 Although the immediate benefits of stricture treatment, such as relieving obstruction and improving biochemistry, are well-established, their long-term implications for TFS remain somewhat obscure. Evaluating long-term outcomes remains challenging, especially given the difficulty of conducting randomized trials to answer this question. Model-based comparisons indicating a potential survival benefit remain a topic of debate.233,238 PSC progression involves intermittent major episodes of obstruction and inflammation, contributing to the deterioration of liver function and accelerating the progression to cirrhosis. Some medical centers advocate for scheduled prophylactic dilation of HGS to prevent complications. Indeed, in a relevant study, 268 patients with HGS were offered the option of either scheduled ERCP dilations at predefined intervals or dilations performed only in response to clinical symptoms or complications. The results revealed that 51% of patients undergoing scheduled ERCP remained transplant-free at 5 years, in contrast to 29% in the clinically indicated ERCP group.239 Despite these findings, most centers do not currently adopt scheduled procedures. Thus, the evaluation of different endoscopic approaches for treating dominant strictures in PSC reveals heterogeneous outcomes (Table 4).235 Studies investigating interventions, including balloon dilation and stent placement, have demonstrated varying overall effectiveness. However, analyses comparing balloon dilation to stent placement indicate potential advantages of balloon dilation, as it may be associated with lower complication rates, suggesting that among the available options, balloon dilation could be considered the primary choice for the initial treatment of PSC-related dominant strictures.235
Studies on endoscopic therapies for dominant strictures in PSC.
| Study | Interventions | Patients | Outcomes | Conclusions |
|---|---|---|---|---|
| 1 | Balloon DilationFerreira, 2021 | 467 | - No significant differences in clinical efficacy and transplant rates.- Lower risk of adverse events with balloon dilation. | - Balloon dilation is the initial treatment of choice for dominant strictures in PSC. |
| 2 | Repeated Balloon DilationGotthardt, 2010 | 96 | - Complications: pancreatitis (2.2%), bacteremia (1.4%), bile duct perforation (0.2%).- Transplant-free survival: 81% at 5 years, 52% at 10 years. | - Repeated balloon dilations effectively preserve bile duct function for many years. |
| 3 | Dilation vs. Dilation + StentingKaya, M. 2001 | 71 | - No obvious benefit from stenting after balloon dilation.- More complications with stenting (45% vs. 6.7%). | - Stenting is not routinely indicated after dilation for dominant strictures in PSC. |
| 4 | 20-Year Experience with Endoscopic TherapyPonsioen 2018 | 117 | - Complications: 7.3%, no procedure-related deaths.- Observed survival at 3 and 4 years higher than predicted by Mayo Clinic model. | - Endoscopic therapy benefits patients with deteriorating clinical courses, with acceptable complications and higher-than-expected survival. |
| 5 | Ursodeoxycholic Acid and Endoscopic TreatmentGluck, 2008 | 106 | - Effective balloon dilations.- Transplant-free survival at 5 years: 100% (Stage 2), 72% (Stage 3), 50% (Stage 4). | - Endoscopic opening of dominant strictures is effective and adds value to medical treatment. |
| 6 | Balloon Dilation vs. Short-term StentsStiehl, 2002 | 65 | - Similar cumulative recurrence rates (0.34 with stent, 0.30 with dilation at 24 months).- More severe adverse events with stents (45% vs. 6.7%). | - Balloon dilation should be the initial treatment for dominant strictures in PSC, particularly for patients with an intact papilla. |
A clinical scenario that warrants particular attention involves long, rapidly progressive, or very tight strictures, as these features should raise suspicion for CCA and prompt further evaluation with ERCP, including ductal sampling via brush cytology and/or endobiliary tissue acquisition for histopathological assessment.3,235 A 25-year longitudinal single-center experience from United Kingdom on 128 patients with PSC suggested worse survival in patients with HGS (13.7 years vs 23 years), with much of survival difference attributable to a significant higher risk of CCA in patients with HGS, with half of those CCA presenting within 4 months of diagnosis of HGS, thus reinforcing the importance of an exhaustive evaluation of a new HGS.240
Historically, brush cytology, the preferred diagnostic tool for evaluating HGS, has been hindered by its low sensitivity. While it remains a cornerstone in the diagnosis of CCA during ERCP, its widespread use is tempered by its suboptimal sensitivity.3,235 A recent metanalysis of 11 studies including 747 patients showed a pooled sensitivity and specificity of 43% and 97%, of bile duct brushing for the diagnosis of CCA, respectively.241 In fact, the literature reports a wide range of accuracy (8–85%) of bile duct brushing in this scenario.242,243 This poor sensitivity has fostered the development of newer techniques to increase diagnostic accuracy, such as DNA aberrations measured by FISH analysis, digital image analysis, flow cytometry, and next-generation sequencing (NGS) markers.3 A Mayo Clinic study involving 235 PSC patients with HGS found that 51% were FISH positive, but only 35 were confirmed to have CCA on histopathological evaluation.244 The study suggests that in PSC patients, the coexistence of HGS and FISH polysomy provides a specificity of 88% for CCA, although the sensitivity remains relatively low at 46%. Conversely, FISH trisomy/tetrasomy showed lower accuracy, with a sensitivity of 25% and specificity of 67%.244 In a subsequent meta-analysis of 8 studies including 828 patients the pooled sensitivity and specificity of FISH for the diagnosis of CCA were 68% and 70% respectively, whereas those for FISH polysomy were 51% and 93%, respectively.245 These data support the high specificity (88–93%) of FISH polysomy for CCA diagnosis in patients with PSC, but with low sensitivity (46–51%). Advances in NGS for cancer diagnostics have introduced a promising combination: a 28-gene NGS panel (BiliSeq) used in combination with pathological evaluation. This approach yielded 83% sensitivity and 99% specificity for diagnosing malignant strictures in PSC patients.246 BiliSeq notably improved the sensitivity of biliary brushings for malignancy diagnosis from 35% to 77% and biliary biopsies from 52% to 83%.246
Beyond brush cytology, a significant breakthrough in the diagnosis of indeterminate biliary strictures was marked by the introduction of single-operator cholangioscopy (SOC), which allows for the direct examination of the biliary tract mucosa and vascular pattern, facilitating the acquisition of samples using biopsy forceps.3,199 A meta-analysis in 2016, including 21 studies, demonstrated that among all ERCP-based interventions (cytology, FISH, confocal laser endomicroscopy), SOC with targeted biopsies is the most accurate modality for diagnosing CCA in patients with PSC, with a diagnostic accuracy of 96% and a pooled sensitivity and specificity of 65% and 97%, respectively.247
In contrast, outcomes from a prospective study conducted in Sweden with 47 patients diagnosed with PSC demonstrated the limited ability of SOC to differentiate benign from malignant strictures.248 However, the introduction of narrow-band imaging (NBI) during SOC yielded a notable 48% increase in the number of suspicious lesions biopsied, facilitated superior visualization of tumor margins. However, NBI-directed biopsies did not increase in the dysplasia detection rate.249 Techniques of potential but still unknown utility include probe-based confocal laser endomicroscopy (pCLE) and endoscopic ultrasound with fine-needle aspiration (USE-FNA). pCLE has shown promising results with high sensitivity in a small series, but evidence for its diagnostic accuracy and availability in clinical practice remains limited.3 In a prospective study with 59 PSC patients presenting with dominant strictures, pCLE demonstrated high sensitivity (85.7%) and specificity (73.1%) for detecting CCA. Notably, sensitivity varied by stricture location, being highest at the bifurcation (100%) and right hepatic duct (100%) and lower at the common bile duct (25%) and left hepatic duct (28.6%).250 On the other hand, evidence supporting the efficacy of USE-FNA for the detection of CCA in PSC remains limited.
Is it necessary to rule out the presence of IBD in patients with PSC? How often?Recommendations:
- •
For patients without IBD, ileocolonoscopy with biopsies from all segments, including the terminal ileum, irrespective of the presence of lesions, is recommended at the time of PSC diagnosis. (LoE 3, strong recommendation).
- •
In patients without IBD, diagnostic ileocolonoscopy can be considered at five-year intervals or whenever symptoms indicative of IBD arise. (LoE 5, weak recommendation).
A strong association exists between PSC and IBD, with an estimated prevalence ranging from 50% to 85% in patients with PSC. Among patients with IBD, approximately 80% have ulcerative colitis (UC), 15% have Crohn's disease (CD), and 5% are diagnosed with indeterminate colitis (IC). The overall prevalence demonstrates geographical variability, with the highest rates reported in Northern Europe, reaching approximately 80%.251,252
PSC-IBD presents with a distinctive phenotype characterized by a high prevalence of pancolitis, with more pronounced inflammation predominantly observed in the right colon, backwash ileitis, and minimal rectal involvement. Notably, there is a considerable risk of colorectal cancer development in these individuals.251,253–256 Following ileoanal anastomosis colectomy, there is an increased propensity for the development of pouchitis in patients with PSC,257 whereas individuals with PH are at an elevated risk for stomal and peristomal varices.258 PSC-IBD may be asymptomatic even in the presence of endoscopic or histological activity. In fact, it is not uncommon to find histological activity in the absence of visible mucosal lesions on endoscopy.251,253,259
The temporal relationship between PSC and IBD is variable, either disease may appear first, and in some cases, IBD may even develop after LT.217 Given the often-subtle clinical presentation and increased risk of colorectal cancer, ileocolonoscopy with systematic biopsies from all colonic segments remains critical for the diagnosis of IBD in patients with PSC, especially after PSC diagnosis, even in the absence of visible endoscopic changes.3,193,235
If IBD is not identified after PSC diagnosis, considering the risk of development at any point during its clinical course, ileocolonoscopy with biopsies should be considered at intervals of five years or whenever clinical suspicion of IBD arises.3,193,235
Fecal calprotectin (fCAL) is a non-invasive biomarker for intestinal inflammation that is widely used in clinical practice. It positively correlates with intestinal permeability to neutrophils and colitis. As an excellent diagnostic tool for IBD, fCAL effectively monitors inflammatory activity owing to its high predictive value for clinical recurrence. However, data regarding the use of fCAL in PSC-IBD are limited. In a small adult series, the correlation between fCAL and ulcerative colitis endoscopic index of severity (UCEIS) was weak in patients with PSC-IBD. Furthermore, patients with endoscopically inactive colitis had higher fCAL than patients with UC. This suggests that fCAL in PSC-IBD may not originate solely from the intestine.260,261 However, in a pediatric series, fCAL in patients with PSC-IBD has shown excellent correlation with mucosal healing, similar to that of UC patients without PSC.259 Although calprotectin may be useful for monitoring patients with PSC-IBD, more data are needed to recommend its use, at least in adults.
Is it necessary to screen patients with PSC for hepatobiliary neoplasms and colorectal cancer? How frequently?Recommendations:
- •
It is recommended that adult patients with large duct disease undergo regular surveillance for CCA and gallbladder malignancy at least annually. This should be conducted regardless of the disease stage, using abdominal imaging, preferably MRI/MRCP. Surveillance is considered unnecessary for patients with PSC who are under 18 years of age, or for those with small-duct PSC (LoE 3, weak recommendation).
- •
Semi-annual monitoring using abdominal ultrasound is recommended for patients diagnosed with PSC and gallbladder polyps measuring<8mm. Cholecystectomy should be considered for patients with polyps>8mm or those demonstrating an increase in size, due to the increased risk of dysplasia or cancer. (LoE 3, strong recommendation).
- •
Semi-annual screening using abdominal ultrasound is recommended for HCC surveillance in patients with cirrhosis. (LoE 3, strong recommendation).
- •
For patients with PSC-IBD aged≥15 years, annual surveillance colonoscopy with targeted biopsies employing dye-based chromoendoscopy is advised (or every 1–2 years in cases with individualized considerations lacking inflammatory activity), regardless of the duration of IBD or liver transplant status. (LoE 3, strong recommendation).
- •
Measuring CA19.9 levels is not recommended in the surveillance of patients with PSC due to its high variability, which results in inadequate precision. (LoE 3, weak recommendation).
- •
CA19.9 levels can be evaluated in cases where CCA is suspected, brush cytology and/or histology findings are inconclusive. (LoE 3, weak recommendation).
PSC is linked to a markedly elevated risk of CCA, 160–400 times higher than that in the general population.262 CCA can develop at any stage of the clinical course of PSC, although the first year after diagnosis carries the highest incidence, accounting for 2.5% of cases and up to 30–50% of all CCA associated with the disease. In subsequent years, the annual incidence is estimated to range between 0.6% and 1.5%.262–264 The cumulative risk of CCA at 10, 20, and 30 years is 6%, 14%, and 20%, respectively.262 Moreover, patients with PSC-IBD exhibit a 28-fold increased risk of CCA compared to those with IBD alone.265
Advanced age is the most consistent risk factor for CCA, with rare diagnoses occurring in pediatric patients or those with small-duct PSC. Additional risk factors include male sex, the presence of HGS, prolonged duration of UC, and advanced liver disease, as indicated by a Mayo score>4, elevated bilirubin levels, and variceal bleeding. Tobacco and alcohol consumption may also contribute to this CCA risk.262,264–266
Although the risk of HCC is higher in patients with cirrhosis, it remains lower than that of CCA. Notably, a 2.4% HCC incidence rate was reported in a 10-year follow-up study.267
Differentiating the clinical presentation of early stage CCA complicating PSC may be challenging, as it is often asymptomatic. Suspicious symptoms and signs of CCA, including rapid deterioration of liver function, abdominal pain, weight loss, jaundice, and persistent elevation of CA19.9, warrant further investigation. Moreover, the clinical presentation of early stage CCA may mimic that of PSC, further complicating its diagnosis.242
Despite the well-documented increased risk of CCA and its impact on the natural history of PSC, there is a notable lack of evidence regarding the surveillance strategies for hepatobiliary malignancies in patients with PSC.3 The heterogeneity of the disease and the limited data regarding the impact of surveillance on survival complicates the development of universal screening strategies. Consequently, risk stratification and the development of individualized surveillance protocols are warranted.3 Although no prospective studies have been conducted, data from a large cohort study showed that regular surveillance was associated with a significantly higher 5-year survival rate (68% vs. 20%, p<0.001).267
The diagnosis of CCA in patients with multiple strictures poses significant challenges. Typically, these malignancies locally invade the hilum and progress longitudinally within the common bile duct, with only a minority presenting as an intrahepatic mass.240,263 Screening for CCA may involve imaging tests alone or in conjunction with tumor markers. While MRI/MRCP has demonstrated superiority over abdominal ultrasound in the early detection of CCA, both modalities, along with contrast-enhanced CT, exhibit limited sensitivity and specificity for diagnosis.3 Ultrasound has been reported to yield a sensitivity of 57% and specificity of 94%, whereas MRI/MRCP demonstrates a sensitivity of 89% and specificity of 75%. However, considerations such as limited availability, cost, and risk of false positives necessitate careful evaluation when utilizing MRI/MRCP. Despite these challenges, annual surveillance using MRI/MRCP is recommended.3,193,267 The recently proposed diagnostic criteria for hilar CCA using MRI/MRCP include the presence of a mass or perihilar soft tissue thickening with progressive enhancement in the late phase. However, these criteria have not yet been validated in patients with PSC.234,268
Positron Emission Tomography (PET) has been proposed as an adjunctive imaging modality. However, its routine utilization for CCA diagnosis is not recommended due to its low specificity and sensitivity, particularly in PSC. In cases with infiltrative morphologies, which are more commonly observed in PSC, the specificity of PET imaging decreases to 18%, whereas it increases to 85% in lesions that exhibit a mass effect. Furthermore, false positives have also been linked to inflammation and bacterial cholangitis.269,270 The utility of PET imaging appears to be greater in advanced stages of the disease, for staging purposes,88 and in the assessment of post-surgical recurrence.272
Among serum tumor markers, CA19-9 is the primary tumor marker associated with CCA. However, its considerable interindividual variability, partly genetically determined, challenges the establishing a cutoff point with sufficient sensitivity and specificity.273–275 Additionally, CA 19-9 levels are influenced by the presence of HGS or bacterial cholangitis, occasionally reaching values like those observed in CCA patients in patients with acute biliary infections. Nevertheless, the intraindividual variability of CA19-9 is minimal, thereby persistently low levels, potentially suggesting the absence of CCA as a complication. Conversely, in the absence of bacterial cholangitis, alterations or progressive elevations of CA19-9 levels warrant further investigations to exclude CCA.276 In a large retrospective series incorporating CA19-9 into the surveillance program, it emerged as an independent factor for survival in patients with CCA. However, because all tumors in this study exhibited suspicious signs on imaging tests, CA19-9 may serve as an additional tool in cases of diagnostic uncertainty rather than for routine surveillance purposes.267 In combination, it has been observed that a CA19-9 cutoff of 20YU/ml (within the normal range) combined with MRI/MRCP or ERCP achieved a sensitivity of 100%, albeit with low specificity for both techniques, 38% and 43%, respectively.242,277 Considering the available evidence and the limited specificity of CA19-9, annual surveillance with MRI/MRCP is recommended.3,193
Patients with PSC have a 9–78-fold increased risk of gallbladder cancer compared to the general population.277 In fact, gallbladder polyps have been identified in 6%-16% of patients with PSC, representing a premalignant condition. The risk of malignancy increases with polyp size, with a lower risk observed in polyps<5mm.278 In contrast, using a size threshold of >8mm on ultrasound to predict malignancy in gallbladder polyps yields a sensitivity of 96% and a specificity of 53.279 Although the optimal imaging modality for gallbladder cancer surveillance remains undefined, ultrasound offers high sensitivity and specificity for polyp detection.280 As a result, it is widely accepted as a cost-effective tool for semi-annual monitoring of gallbladder polyps smaller than 8mm.193 For polyps>8mm, cholecystectomy should be considered. However, the decision to proceed with surgery or semi-annual surveillance should be based on factors including liver function, risk of perioperative hepatic decompensation, and hepatobiliary infection risk.3,193
In addition to biliary tract malignancies, HCC represents another potential complication in PSC, though it is rare in the absence of cirrhosis.262,281 However, semi-annual ultrasound surveillance is warranted in PSC patients with cirrhosis, as it is in patients with cirrhosis of any other etiology.282
Another neoplasm with increased risk in PSC is colorectal cancer (CRC), with incidence rates reported to be 5–12 times higher than in the general population262,283,284 and 4–5 times higher than that in patients with IBD without PSC.254,255,262,265 A meta-analysis including 1022 patients from 16 studies showed a 3-fold increased risk of dysplasia/CRC in individuals with PSC-IBD compared to those with IBD alone.256 Furthermore, progression from low-grade dysplasia to high-grade dysplasia or CRC occurs more rapidly in PSC-IBD patients than in those with IBD alone.285 The specific risk of CRC in PSC patients without IBD with regards to the general population remains unknown. Additionally, the risk of CRC persists after LT.286 Recent studies have reported an accumulated CRC incidence of 7% and 9% at 5 and 10 years, respectively, with a similar rate observed in children, reaching 5% at 10 years.287 CRC typically appears at a young age and predominantly affects the right colon. Early-onset of IBD is a recognized risk factor for CRC development in patients with PSC-IBD.265,285,287 Notably, children diagnosed with IBD before the age of six exhibit a higher CRC risk compared to those diagnosed during adolescence.287 Chronic inflammation, which is often underestimated in both children and adults, likely contributes to the increased CRC risk.259 Notably, adherence to CRC surveillance guidelines for IBD patients has been associated with lower CRC rates in PSC-IBD patients.288 Consistent with guidelines from the European Society of Gastrointestinal Endoscopy235 (ESGE), European Crohn's and Colitis Organization (ECCO),289 and American College of Gastroenterology (ACG),191 surveillance via colonoscopy with biopsies at intervals of 1–2 years is recommended, starting after PSC-IBD diagnosis and from the age of 15 years.3,193 Targeted biopsies utilizing chromoendoscopy enhance the detection rate compared to standard endoscopy with random biopsies and are, therefore, the preferred technique per ESGE recommendations. Emphasis should be placed on achieving adequate colon preparation to optimize the detection rate.235Fig. 4 shows the recommended follow-up of patients with PSC.
What is the management of infectious cholangitis in patients with PSC? Is it necessary to administer antibiotic prophylaxis to patients with PSC and recurrent cholangitis?
Recommendations:
- •
The treatment of acute bacterial cholangitis requires antibiotic therapy, and if a significant underlying stricture is found, prompt biliary decompression is recommended. (LoE 3, strong recommendation).
- •
In advanced stages of the disease, a prolonged course of rotating antibiotics may be considered in selected patients to reduce the risk of recurrent cholangitis. (LoE 5, weak recommendation).
- •
Recurrent cholangitis should be considered as an indication for LT in case of repeated episodes of cholangitis not controlled with standard therapy. (LoE 4, strong recommendation).
Bacterial cholangitis is a frequent complication of PSC, usually occurring in advanced stages of the disease and often associated with HGS. In this setting, imaging, particularly MRCP, is recommended to assess the extent of biliary obstruction and to guide potential endoscopic or interventional management.190 The clinical presentation of bacterial cholangitis in PSC varies widely, ranging from mild with nonspecific symptoms to severe biliary sepsis. Notably, liver biochemistry may remain unchanged, especially when the infection is limited to smaller intrahepatic ducts. Diagnosis may rely on the response to antibiotic therapy alone, as standard definitions for cholangitis are not universally applicable in PSC.290 In fact, the definition and diagnosis of cholangitis in PSC remain challenging, with new criteria proposed for acute cholangitis in this population yet to be validated in larger cohorts. Additionally, recent efforts by the International PSC Study Group to establish a consensus on these criteria have been inconclusive.197 As per the proposed definition, the diagnosis of acute bacterial cholangitis in PSC necessitates meeting either a single criterion (evidence of suppurative cholangitis on ERCP) or at least one major criterion (body temperature>38°C, leukocyte count>12/ml, or C-reactive protein>75mg/L), along with at least two minor criteria (positive bile culture, elevated ALP or total bilirubin>2× ULN, absence of other infectious foci).276
Despite the ongoing debate about the diagnosis of bacterial cholangitis, Kaplan et al. reported that approximately 6% of patients with PSC present with superimposed bacterial cholangitis at diagnosis, with nearly 40% experiencing this complication over the course of the disease.291 In a phase II clinical trial evaluating simtuzumab, bacterial cholangitis emerged as the most frequent disease-related complication, occurring in 13% of 234 PSC patients during a median follow-up of 23 months.214 The precise impact of bacterial cholangitis on disease progression remains uncertain but warrants further investigation.190
Bile cultures obtained from patients with PSC reveal a wide range of bacterial species, irrespective of prior biliary interventions. However, data on patients without previous ERCP remain limited, as invasive sampling in this population is typically restricted to small cohorts. Bjornsson et al. conducted a study comparing bile cultures from PSC patients and those with other cholestatic disorders, revealing positive cultures in 3 out of 12 ERCP-naive PSC individuals (25%) and 6 out of 10 PSC patients with previous ERCP, but these differences did not reach statistical significance. Among the positive bacterial cultures, 75% consisted of gram-positive organisms, while the remaining 25% were enteric bacteria, a distribution that differed significantly from disease controls with common bile duct stones (74% enteric bacteria and 26% gram-positive bacteria) studied for comparison.292 HGS poses a risk for bacterial colonization due to biliary flow restriction. Positive bacterial cultures were observed in 23 of 37 PSC patients with HGS (62%) compared to only 4 of 13 patients without stenosis. Notably, enteric bacteria were detected in the bile of 19 of 37 PSC patients with HGS (51%), but never in the absence of such stenosis.293 Additionally, portal bacteremia, documented in patients with active colitis, may represent another crucial contributing factor to cholangitis in PSC-IBD.294
Moreover, ERCP is a significant risk factor for bacterial cholangitis in PSC, particularly when involving stent insertion. Post-ERCP bacterial cholangitis is documented in 2–8% of PSC patients undergoing this procedure.244,295 In a multicenter randomized trial targeting individuals with PSC and HGS, short-term stents were associated with higher treatment-related adverse events, including bacterial cholangitis (12% vs. 3%), than balloon dilatation, although no disparities in recurrence-free patency were observed.296 Controversy persists regarding the performance of biliary sphincterotomy/papillotomy in PSC patients due to the associated short-term complications. However, in cases of difficult biliary cannulation or anticipated future ERCP, sphincterotomy is generally recommended in the absence of contraindications, with preference for a small sphincterotomy.235,295 To reduce the risk of post-ERCP cholangitis, peri-interventional antibiotics are advised, unless patients are already receiving antimicrobial therapy. Nonetheless, the selection of the antibiotic agent and the optimal duration of treatment remain unclear.235,297
The presence of HGS and the history of ERCP have also been associated with Candida infections in bile. In an initial investigation conducted in Heidelberg et al., Candida species were isolated from bile samples in 8 out of 67 (12%) individuals with PSC undergoing ERCP, while Aspergillus was not detected. Most patients (7 out of 8) had advanced PSC with HGS, and all had a history of antibiotic treatment and/or ERCP.298 In a subsequent analysis, biliary candidiasis was identified in 30 of 150 (20%) patients. Factors associated with an increased risk of developing biliary candidiasis included age at PSC diagnosis and the number of prior ERCP.299 Furthermore, a specific genotype of fucosyltransferase 2 (FUT2) has been identified as a risk factor for both HGS and biliary Candida infections in PSC.3
In terms of prognosis, a positive bacterial culture of bile in the presence of a HGS was not correlated with a worse prognosis,193 and bacterial cholangitis did not affect survival among patients with PSC awaiting LT, because of successful treatment.300 However, Candida infection may be linked to a significantly poor prognosis.190 In the Heidelberg study, although all patients had similar baseline characteristics, individuals with persistent biliary candidiasis showed reduced TFS rates and a notably increased incidence of CCA.299
Bacterial cholangitis should be treated with antibiotics.3 These infections are usually polymicrobial, and antibiotic selection should be guided by local practice, considering bacterial sensitivity and the degree of liver and/or renal impairment. The most frequently isolated microorganisms include Gram-negative bacteria such as Escherichia coli, Klebsiella, Pseudomonas, and Bacteroides species, as well as Gram-positive Enterococci or Streptococci.301 Consequently, the initial antibiotic choice should provide coverage against both Gram-negative and Gram-positive bacteria. Amoxicillin/clavulanate is a suitable first-line agent for mild episodes owing to their oral administration options. Fluoroquinolones, previously used as first-line agents, should now be reserved for specific cases owing to their high resistance rates and unfavorable side effects, in alignment with antimicrobial stewardship principles. More severe cases warrant intravenous antibiotics such as piperacillin/tazobactam or third-generation cephalosporins with adequate anaerobic coverage.301 In patients with sepsis or those who do not have early response to initial antibiotic therapy while awaiting biliary decompression, additional coverage against gram-positive organisms targeting Enterococci, such as glycopeptide antibiotics (e.g., vancomycin) or oxazolidinone antibiotics (e.g., linezolid), may be considered.301 Due to the uncertain benefit of antifungal therapy for biliary Candida infections, patients are often not treated for fungal infection unless they have an immunosuppressive condition or overt cholangitis.302
Given the frequent association between cholangitis in PSC and HGS, it is advisable to perform an MRCP at diagnosis. Patients with severe acute cholangitis and HGS have an increased risk of mortality and require urgent biliary decompression.302 Short-course antibiotic treatment alone is insufficient to eradicate bacteria from the bile ducts of patients with HGS without endoscopic intervention, but most patients respond to endoscopic drainage of the obstruction combined with antibiotics.302 In patients with milder bacterial cholangitis, it may be feasible to wait longer for a response to antibiotic treatment before proceeding to ERCP and dilatation of strictures, if indicated.302 EASL-ESGE guidelines recommend dilatation of a HGS if it is deemed to be the cause of complications, in this case bacterial cholangitis.235 The decision to perform balloon dilation as the primary option and/or subsequent stenting of a stricture should involve a multidisciplinary care team, including an endoscopist, and should be based on various individual patient considerations. These considerations include the perceived adequacy and durability of the response to balloon dilation and the patient's ability to return for stent removal within an appropriate time frame. When balloon dilation is performed, it should not exceed the diameter of the bile ducts, immediately delineating the stricture. If a plastic biliary stent is placed, it should typically be removed within 4 weeks to minimize the risk of adverse events.302 The role of self-expanding metallic stents in PSC remains unclear; however, their use may be considered in select cases. Repeat therapeutic intervention for a persistent relevant stricture should be done if they are the cause of symptoms and complications.235
Occasionally, patients with recurrent bacterial cholangitis due to complex intrahepatic cholangiopathy may require long-term prophylaxis with antibiotic rotation.190 However, this approach should be considered only under exceptional circumstances because of the associated risk of antibiotic resistance. Evidence regarding the effectiveness of maintenance antibiotic treatment in preventing cholangitis in patients with PSC is limited. Most data come from other conditions where the biliary tree may be compromised, such as after the Kasai procedure for biliary atresia, conditions predisposing to ascending cholangitis, or obstructions from various causes. In long-term antibiotic therapy, the excretion of antibiotics into the bile may be crucial. It has been hypothesized that the threshold concentration of bacteria in bile must be exceeded for clinical signs of cholangitis to manifest. Maintenance therapy with antibiotics may help maintain the concentration of bacteria below this threshold or prevent sepsis in the event of bacterial translocation into the systemic circulation.303 The most appropriate antibiotics for maintenance therapy must be active against the most common biliary pathogens, have low toxicity even after prolonged use, be available in an oral preparation at an acceptable cost, and reach sufficient levels in the bile. Trimethoprim-sulfamethoxazole (TMP-SMZ) meets these criteria, especially after repeated dosages.303 The optimal duration of antibiotic therapy is unknown. Typically, patients are treated for 3–4 months, and if cholangitis recurs, maintenance therapy should be reinstated. Fluoroquinolones can be used as alternatives to TMP-SMZ for maintenance therapy.303 Another alternative is the combination of amoxicillin and a β-lactamase inhibitor whose biliary excretion is lower, but in an unobstructed biliary tree, it still reaches therapeutic levels.303
Recurrent cholangitis can reach such a severity that it becomes the primary indication for LT when repeated episodes are not controlled by antibiotics.304 There is considerable international variation in practice, with 17% of PSC patients transplanted for this indication in Norway, while less than 5% are listed for this indication in the UK or United States.304 However, a study from two US transplant centers showed that transplant candidates with PSC and bacterial cholangitis did not have an increased risk of waitlist mortality, which raised questions about whether recurrent cholangitis should be an accepted indication for LT.304 Analysis of UNOS outcomes suggests that patients with PSC listed for LT have lower mortality and are less likely to be removed from the list than patients listed for other indications, despite similar MELD scores, although there are conflicting results, as detailed in LT for PSC section. This indicates that MELD may overestimate the severity of liver dysfunction in PSC patients. Under specific circumstances, PSC patients with documented non-iatrogenic recurrent bacterial cholangitis may receive additional MELD points, thereby obtaining higher waiting list priority.305 MELD exception points can be granted for recurrent cholangitis with more than two episodes of bacteremia or more than one episode of septic complications within a certain timeframe (e.g., 6–12 months).305
Is UDCA useful in the treatment of PSC?Recommendations:
- •
UDCA may be used for PSC treatment at a dose of 15–23mg/kg/day. (LoE 1, weak recommendation).
- •
The use of low-dose UDCA may be considered to reduce the risk of colorectal cancer in patients with IBD, although it does not appear to provide protection against CCA. (LoE1, weak recommendation).
UDCA is the most widely used pharmacological treatment for PSC.275,306 Treatment with UDCA has demonstrated improvement in biochemical parameters, a variable impact on cholestatic symptoms, and no influence on TFS, likely due to the limited statistical power of available studies.306 The only study suggesting the benefit of UDCA on TFS was a Japanese retrospective study307 that included 325 patients with PSC, with a median follow-up of 5.1 years. During this period, 57 patients died and 24 underwent LT. In Cox regression analysis, UDCA use was associated with longer TFS (aHR 0.47). However, no association was observed with overall survival or all-cause mortality. Notably, the study did not report treatment duration or UDCA dose.307 In contrast, a meta-analysis including 553 patients concluded that UDCA does not reduce mortality risk compared to placebo (RR 1.04) or the need of LT (RR 1.22). Despite improvements in biochemical parameters, no histological benefit, reduction in CCA incidence, or symptom improvement was found.308 Furthermore, these conclusions are consistent with the 2003 Cochrane review.309 The dosage of UDCA is critical, doses below 15mg/kg/day improve the biochemical profile without significantly reducing disease progression, whereas high doses (28–30mg/kg/day) are harmful. In a controlled, double-blind, randomized study, patients receiving high doses had worse outcomes, including more decompensation, need for transplantation, and death, despite biochemical improvements.280,310
A Chinese meta-analysis indicated that the combination of UDCA with metronidazole (MTZ) demonstrated superior efficacy compared to UDCA alone or in conjunction with mycophenolate mofetil (MMF) in terms of survival rates and the impact on histological progression. However, this combination was also associated with an increased incidence of side effects.311 Treatment with UDCA resulted in a significant reduction in bilirubin and ALP levels compared to the untreated group and the combination of UDCA and MTZ showed histological benefit, although not significantly different from the other comparison groups.311
However, the evaluation of response and its association with relevant clinical endpoints is challenging, as it is the disease itself. In 2016, the PSC Study Group suggested that ALP, bilirubin, LSM, and histology could be acceptable potential surrogate endpoints to evaluate the impact of treatments.203 Regarding the benefit of UDCA on biochemical surrogates or symptoms, it has been shown that withdrawal of UDCA leads to increased pruritus, ALP levels, and deterioration of PSC Mayo Score, as referred to in a study of 26 PSC patients who had received UDCA at stable doses of 12–15mg/kg/day. Serum bile acid profiling before and after UDCA withdrawal revealed a significant reduction in lithocholic acid and its derivatives after discontinuation, with no notable changes in primary bile acid concentrations, except for the increased accumulation of their taurine conjugates.312
Norucholic acid has shown benefit in animal models of cholangitis and biliary fibrosis, with regression of sclerosing cholangitis after 4 weeks of treatment.313 Preliminary findings presented at the EASL meeting in 2025 indicated that, after 96 weeks of treatment, a significantly higher proportion of patients receiving norucholic, compared to those receiving a placebo, exhibited improvements in ALP and liver histology. Specifically, 15.1% of patients treated with norucholic achieved the combined primary endpoint, defined as an improvement in ALP to <1.5× ULN without a worsening of fibrosis stage at week 96, compared to 4.2% of those in the placebo group.314 The results from the phase II trial comparing elafibranor with placebo have also been published. Patients were randomized to receive elafibranor 80mg, 120mg, or placebo for 12 weeks. Both elafibranor doses led to significant reductions in ALP compared to placebo, with ALP normalization observed only in the treatment arms (9.1% and 17.4%, respectively). The safety profile was favorable, as serious treatment-emergent adverse events occurred exclusively in the placebo group.315
Altogether, although there is no evidence for the use of UDCA as a modifier of the natural history of PSC, the available data on the impact on ALP as a surrogate marker of overall disease progression has led the EASL3 to recommend treatment at doses of 15–20mg/kg/day. Similarly, the AASLD has modified its recommendations, suggesting initiating treatment with UDCA at doses of 13–23mg/kg/day and maintaining it if there is good tolerance and improvement in ALP or symptoms after one year of treatment.193
Apart from the benefit on the natural history or symptoms of PSC, it is important to comment on the benefit of UDCA on PSC-associated IBD, or the risk of developing CCA or CRC both increased in PSC patients with IBD compared to the general population and IBD patients without PSC. The predominance of CRC in the right colon of PSC patients has been related to greater mucosal exposure in these areas to excess unabsorbed secondary BA in the intestine.316 Carcinogenesis in these patients is different from that in sporadic CRC, where most cases follow the adenoma-carcinoma sequence. In IBD associated with PSC, there is a sequence of genetic alterations related to genomic instability induced by chronic inflammation, leading to neoplastic changes.316,317 Secondary BA in feces, including deoxycholic and lithocholic acids, have been linked to the pathogenesis of colorectal cancer by altering the balance between crypt cell proliferation, differentiation, and apoptosis.317 Elevated levels of lithocholic acid are presumably produced by the 7-dehydroxylation that intestinal bacteria perform on unabsorbed BA that pass into the colon. Regarding the utility of UDCA as a chemoprotective agent against cancer, animal models have suggested that UDCA decreases colon carcinogenesis, and it has been shown that UDCA reduces deoxycholic acid concentration.318,319
In humans, two systematic reviews and meta-analyses320,321 found no association between UDCA use and CCR risk in IBD. However, both studies reported a potential protective effect in a subgroup of patients treated with low-dose UDCA. The first of these publications is a systematic review and meta-analysis of clinical trials, case-control studies, and cohort studies, but highlights the heterogeneity of UDCA doses among studies (ranging from 8mg/kg/day to 30mg/kg/day) and points toward the benefit in the reduction of cancer risk in studies evaluating low or moderate doses of UDCA.320 In the second systematic review and meta-analysis,321 763 patients with IBD and PSC were included, of whom 117 were diagnosed with CRC. The data were derived from eight studies, comprising five observational studies and three randomized controlled trials. Although overall there was no protective association between UDCA and CRC, there was a significant chemopreventive effect on the risk of CRC or high-grade dysplasia (OR 0.35). In the subgroup patients receiving low doses of UDCA (8–15mg/kg/day), a significant reduction in the risk of CRC was recorded (OR 0.19). Based on these results, the authors concluded that UDCA, particularly at low doses, may reduce the risk of advanced neoplasia. Consequently, the American Gastroenterological Association (AGA)322 and the European Crohn's and Colitis Organization (ECCO)223,323 recommend the administration of UDCA in patients with long-standing PSC and IBD.
In contrast to the potential benefit of UDCA in reducing the risk of CCR, a systematic review and meta-analysis224,308 including 3 clinical trials found no evidence of reduced risk of CCA in patients treated with UDCA. However, these findings are challenged by those reported in two studies. A study conducted in a Nordic population, where UDCA treatment decreased the incidence of CCA in patients on the LT waiting list226,324 and a prospective observational French study226,325 including 150 patients, which reported a low incidence (0.38/100 patient-years) of CCA, concluding that these data are indicate a potential chemoprotective effect of UDCA on the development of biliary tract neoplasia.
Are there other effective drugs for the management of PSC?Recommendations:
- •
The use of corticosteroids, antiproliferatives (such as azathioprine or mycophenolate mofetil), methotrexate, or colchicine is not for the treatment, as they have not demonstrated a favorable therapeutic effect. (LoE 4, weak recommendation).
- •
PSC patients with AIH features should be managed in accordance with the guidelines for AIH. (LoE 3, strong recommendation).
- •
Biological drugs (anti-TNF-α and immunomodulators) are not recommended for the treatment of PSC, except in specific cases where they are indicated to control inflammatory bowel disease. (LoE 4, strong recommendation).
- •
The use of antibiotics outside the management of infectious complications cannot be recommended at this moment. (LoE3, weak recommendation).
Immunosuppressants and immunomodulators, including corticosteroids, calcineurin inhibitors, methotrexate, azathioprine, and anti-tumor necrosis factor (TNF) treatments, have not demonstrated benefit in PSC.326–331 Although the exact pathogenesis of PSC is unknown, its association with IBD raises the question of whether therapies targeting IBD positively influence the course of PSC.
Several studies have assessed the effect of anti-TNF-α agents on PSC progression. Infliximab, was evaluated in a double-blind, randomized controlled trial,330 which was terminated early due to futility. Adalimumab has shown a greater reduction in ALP levels than infliximab or vedolizumab in retrospective studies,329,332 although without improvement in LSM or MRCP findings. Hedin et al.,332 in a multicenter study involving 20 tertiary hospitals in the USA and Europe, found that only the adalimumab group showed ALP improvement, with a median reduction of 15% after 3 months of treatment, that was maintained at 6 and 12 months. Vedolizumab, however, showed no impact on ALP levels in various retrospective studies of IBD,332,333 except in patients with PSC and cirrhotic disease, as noted by Lynch et al.304 Simtuzumab, an anti-LOXL2 monoclonal antibody, showed no effect on ALP or fibrosis estimated by ELF, in a phase II clinical trial over 96 weeks.214
Beyond efficacy, the safety profile of these drugs is critical. Infliximab has been linked to ALT increase, nodular hyperplasia, cholestasis, hepatitis B reactivation, and bacterial cholangitis in IBD patients with PSC. A retrospective study in PSC-IBD patients published in 2021 published in 2021155comparing anti-TNF-α therapy to immunomodulators found a higher risk of bacterial cholangitis among patients receiving anti-TNF-α agents than among those treated with immunomodulators (OR 7.29), with an incidence of 31% vs 11.1%, respectively. Additionally, the anti-TNF-α group experienced shorter intervals between episodes of cholangitis. Patients on vedolizumab did not show a significant risk of cholangitis, although the limited number of vedolizumab patients should be acknowledged. These findings suggest the need for further studies to determine whether escalating aminosalicylates to immunomodulators, rather than biological drugs, is more appropriate for patients with PSC, especially those with recurrent cholangitis.334
AntibioticsSeveral clinical and preclinical studies have established the gastrointestinal microbiome alterations associated with intestinal permeability changes as pathogenic factors in PSC.335–338 The relationship between the microbiome and the altered bile acid pool in PSC is bidirectional; the microbiome influences bile acid composition, while changes in the bile acid pool can, in turn, reshape the intestinal microbiome.339 Additionally, the intestinal microbiome in PSC is linked to hepatic innate immune system activation, leading to inflammation and fibrosis.340 Therefore, modifying the intestinal microbiome using antibiotics or fecal transplantation has been proposed as a therapeutic alternative, with several clinical trials and case series available.
Oral vancomycin, a minimally absorbed antibiotic with immunomodulatory effects by reducing cytokine production from T cells,341 has shown effects on ALP levels, PSC Mayo Score, and clinical improvement in various open-label studies342 and randomized clinical trials compared to MTZ343 or placebo.344 Compared to MTZ,343 vancomycin demonstrated better clinical tolerance and has been suggested to prevent disease recurrence after LT.345 Recently, in a single-arm interventional study exploring mucosal changes in PSC-IBD patients, a 4-week course of oral vancomycin induced clinical remission in 12 of 15 patients with PSC-IBD and significantly reduced fecal calprotectin levels. Treatment was associated with distinct microbial and host transcriptomic shifts, including altered bile acid and short-chain fatty acid pathways. Notably, disease activity and mucosal profiles returned toward baseline after treatment cessation.346 Rifaximin, showed no significant changes in ALP, secondary endpoints, or quality of life questionnaires in an open pilot study347 with 16 patients treated for 12 weeks. Minocycline, a second-generation tetracycline tested in an open study on 16 PSC patients treated at different doses for 12 months, exhibited good clinical tolerance and a significant reduction in ALP and Mayo Risk Scores.348
Fecal transplantationSeveral case reports and pilot studies have explored the potential role of fecal microbiota transplantation (FMT) in PSC-IBD based on the premise that FMT enhances microbial diversity, promotes epithelial integrity maintenance, and reduces mucosal permeability and inflammation. In an open, uncontrolled pilot study, Allegretti et al.,349 evaluated the safety, changes in cholestasis profile and microbiota, and metabolomic profile in 10 PSC patients after FMT, showing improvement in IBD-related symptoms and liver biochemistry, which correlated with increased microbiome diversity. There were no changes in the bile acid profile, which, according to the authors, raises the question of whether fecal transplantation acts through microbiota-metabolomic interactions involving metabolites other than BA.
StatinsStatins have antioxidant, antiproliferative, and anti-inflammatory effects, which improve endothelial function and stimulate angiogenesis.350 Although no specific studies on statins in PSC have been published, a Swedish study351 reported that 13.9% of PSC patients received statins, which were associated with a significant reduction in all-cause mortality risk (HR, 0.68), and a 50% reduction in the risk of death or transplant (HR 0.50) and liver-related adverse events (HR, 0.53).
Other drugs- -
Probiotics: In a placebo-controlled clinical trial, no significant changes were observed in clinical outcomes or laboratory data.352
- -
Antifibrotics: Given that PSC is a fibro-obliterative disease of the bile ducts, various clinical trials of antifibrotic drugs have been conducted, mostly yielding negative results.175
- -
Other: Obeticholic acid also showed significant improvements in ALP levels and histological parameters in a phase II clinical trial including 76 patients.353 Similarly, other FXR agonists, such as GS-9674, produced substantial reductions in ALP in a phase II study with 62 participants.354 Additionally, PX-104, a selective FXR agonist (PFXR), demonstrated comparable reductions in ALP in a phase II trial involving 60 PSC patients.355
Recommendations:
- •
The presence of significant biliary obstruction should be rule-out in patients with large-duct PSC and progressive pruritus. If significant stenosis is identified and accessible, it should be managed as previously described. (LoE 4, strong recommendation).
- •
Bezafibrate should be considered as first-line pharmacological treatment management of pruritus. Rifampicin may be used as an alternative in cases where bezafibrate is contraindicated. If neither option is feasible, the treatment strategy is similar with the approach used in PBC. (LoE 4, strong recommendation).
The prevalence of pruritus in PSC ranges between 30% and 60% and significantly impacts the quality of life. Although pruritus may arise even in the absence of significant biliary obstruction, the presence of potentially treatable stenosis or obstruction should always be rule-out.3,193
The exact pathophysiology of pruritus associated with cholestasis is not well understood and is likely to be multifactorial,151,353 as explained in the PBC section. In fact, except for linerixibat -an agent that has not been tested for the treatment of pruritus in PSC-, the management of pruritus is like that in patients with PBC (Fig. 3). The latest drugs incorporated into the treatment of pruritus in PSC are IBAT. However, at the time of this publication, no results from phase III trials are available regarding the efficacy and safety of this therapy in the context of PSC.
When is LT indicated for patients with PSC? How do we manage these patients?Recommendations:
- •
LT should be considered in PSC under the same circumstances, limits, and prioritization system as in other causes of liver disease (decompensated cirrhosis, liver failure, or HCC). (LoE 3, strong recommendation).
- •
LT for PSC should be considered in selected cases of recurrent bacterial cholangitis or intractable pruritus. In these instances, the prioritization system is regarded as an “exception to the MELD score.” (LoE 3, strong recommendation).
- •
High grade dysplasia (HGD) demonstrated through cytology or biopsy may serve as a criterion for considering LT, particularly in the absence of other indications for transplantation. (LoE 4, week recommendation).
- •
Non-resectable hilar CCA may be considered an indication for LT. The indication, management, and prioritization in these cases should align with the current staging criteria and neoadjuvant approaches as per national and international guidelines and consensuses. (LoE4, weak recommendation).
- •
Immunosuppressive (IS) regimens based on tacrolimus are suggested. (LoE 2, weak recommendation).
- •
Duct-to-duct anastomosis should be used as a biliary reconstruction technique in LT for PSC, when feasible and technically possible. (LoE4, strong recommendation).
LT represents a definitive therapeutic option for patients with end-stage liver disease in the setting of irreversible hepatic dysfunction. PSC may lead to various circumstances compromising survival or quality of life, warranting consideration of LT, from advanced cirrhosis or HCC development to more specific situations, such as recurrent bacterial cholangitis or uncontrolled pruritus. CCA or HGD in selected patients is also considered an indication for LT under certain circumstances.304,356,357 The indication for LT in patients with PSC varies across regions, reflecting differences in disease prevalence, the relative burden of other liver disease etiologies, and the availability of grafts. PSC accounts for up to 4–5% of LT indications in Europe and North America,358,359 with the highest reported indication rates in Nordic countries.360
According to the European Liver Transplant Registry (ELRT),305 analyzing LT cohorts across different periods (1980–1989, n=159; 1990–1999, n=1282; 2000–2009, n=2316; 2010–2017, n=2549), there is a growing trend in LT indications for PSC, with a more pronounced increase observed among women, rising from 1.8% in the earliest cohort to 4.3% in the most recent.358 Graft survival following LT in the European registry cohort at 1, 5, 10, 15, 20, and 30 years was 83.6%, 70.8%, 57.7%, 44.9%, 30.8%, and 11.6%, respectively.305 Notably, there were no differences in 10-year graft survival between LT performed in two different historical periods (1990–1994 vs. 2005–2009): 58% vs. 59%.305 In the Scandinavian cohort, Brandsaeter et al.361 found no differences in 1, 3, and 5-year survival compared to other transplant indications. In PSC patients, prior hepatobiliary surgery and MELD score were identified as survival predictors.361
The main determinants of post-LT outcomes in PSC are the development of biliary strictures, rejection, and PSC recurrence which occurs in 20–30% of patients within 10 years following LT. This recurrence is associated with a 4-fold increased risk of graft loss or death, and the impact of both outcomes becomes more significant over time.362,363 Re-transplantation remains a therapeutic option in cases of graft failure due to recurrent disease.359
The prioritization system for PSC on the waiting list is controversial and is influenced by LT indications (advanced cirrhosis-MELD score versus specific PSC complications-MELD exceptions). Various retrospective studies have compared mortality or delisting in PSC patients versus those in other etiologies of liver disease, yielding conflicting results. In a study published in 2003,361 the probability of death while awaiting LT was lower in PSC than in other indications (3.4% vs. 7.3%, p=0.003). A retrospective observational study compared 819 patients on the LT waiting list due to PSC with those listed for other etiologies. Compared to alcohol-related liver disease, the risk of mortality within the first 90 days on the waiting list was similar for HCV and PSC. However, disease progression, estimated by delta MELD-Na, was higher in MAFLD patients.364 While there is consensus on using MELD for cirrhosis and its complications, there is debate over its application in “MELD exceptions” (cholangitis, intractable pruritus, dysplasia),3,193,356 where MELD may confer an advantage over other indications. In its recent consensus conference on LT for PSC, the European Society for Organ Transplantation (ESOT) concluded that the MELD score should be applied to prioritize patients on the LT waiting list for PSC, and in cases of specific PSC complications not reflected in MELD (pruritus affecting quality of life, recurrent bacterial cholangitis, and the presence of HGD), exception points should be considered,3,356 albeit controversial with varying positions from scientific societies. CCA is also included in MELD exceptions, though it requires specific management and considerations.3,193,356
Indications for liver LT in dysplasia and CCAGiven the evidence that dysplasia is the main precursor to invasive CCA,365 the indication for LT in cases of dysplasia over PSC is considered.3,193,356,366 HGD as an indication for LT was initially proposed by Scandinavian groups. Subsequently, considering the strong association between HGD and CCA and the poorer outcomes of LT even in incidental CCA cases,366 the EASL guidelines 3 accepted the finding of HGD in samples obtained by endoscopic brushing as an indication for LT. However, the recent ESOT consensus states that universal recommendations for LT in HGD cases in PSC cannot be made, considering the difficulty in quantifying the risk of HGD development in PSC patients and limited donor pools in many countries. Notably, patients in whom the indication for LT is based on the presence of dysplasia often present severe radiological PSC findings but preserved liver function, framing the listing criteria as “exceptions to MELD”.367
The variability in LT indication criteria for dysplasia reflects the inherent diagnostic challenges, as the inflammatory activity of PSC and changes related to superimposed cholangitis can lead to reactive atypical or dysplastic alterations in brush cytology specimens, complicating the differential diagnosis with true premalignant changes. Various studies have analyzed the concordance between pre-LT cytology data that indicated LT, and explant findings, showing that between 20% and 57% of those transplanted for HGD did not actually have cancer in the explant, thus questioning whether LT decision-making in patients with preneoplastic changes is the most appropriate.368,369 In contrast, in a Scandinavian study that included 209 transplanted PSC patients (none with a previous CCA diagnosis), incidental CCA was diagnosed in 16 patients, nine of whom already had documented dysplasia in the pre-LT study, but in seven cases with incidental CCA, there was no pre-LT dysplasia. In this series, post-LT survival was worse compared to others in cases of incidental CCA, but not in cases of dysplasia.366
Both hilar and intrahepatic CCA have been considered contraindications to LT due to their poor survival outcomes, which are influenced by tumor recurrence. However, in specific situations, LT may be considered; for intrahepatic CCA, it is feasible in unresectable cases, confined to the hepatic parenchyma, and with a small tumor size. Both EASL, AASLD, and SETH contemplate this possibility under research protocols.3,193
For hilar CCA associated with PSC, LT is considered in cases of unresectable disease, early stages, and under strict protocols of neoadjuvant oncological therapy. Patient survival outcomes are better in cases of LT for CCA associated with PSC compared to CCA without prior PSC (5-year survival 74% vs. 58%).370
Immunosuppressive regimensThe role of IS in LT outcomes for PSC remains controversial, particularly regarding its impact on PSC recurrence, patient and graft survival, and IBD progression after transplantation. Current published data are derived from observational studies, limiting the possibility of comparing different IS regimens. Furthermore, these studies present inconsistent results.
A meta-analysis published in 2020371 reported that cyclosporine was associated with lower rates of PSC recurrence and improved survival compared to tacrolimus. However, other studies, including a recent international multicenter cohort involving 531 PSC transplant recipients, found no significant association between the type of calcineurin inhibitor used and the risk of PSC recurrence or post-transplant survival.372,373
A recent study based on data from two major transplant registries, the European Liver Transplant Registry (ELTR) and the Scientific Registry of Transplant Recipients (SRTR), covering the period from 2000 to 2020 and representing most centers in Europe and the USA, employed an intention-to-treat design with propensity score matching. The study demonstrated that starting treatment with tacrolimus, as opposed to cyclosporine, was associated with better patient and graft survivals.374 In detail, after matching, the final cohort included 399 patients treated with cyclosporine and 1197 with tacrolimus, with a median follow-up of 7.4 years. Initial treatment with tacrolimus was superior in terms of patient survival (72.8% vs. 65.2% at 10 years) and graft survival (62.4% and 53.8%, respectively). These findings were consistent across subgroups considering age, sex, registry (ELTR vs. SRTR), period, MELD score, and diabetes. Rejection rates were similar with both immunosuppressive drugs. In multivariate analysis, tacrolimus (HR 0.72) and mycophenolate (HR 0.82) were associated with lower graft loss and death, consistent with findings previously reported in LT for other etiologies.374,375
Graft survival is closely associated with PSC recurrence, which has also been related to development of rejection.376 ESOT in a recent consensus conference recommended an initial triple regimen, ideally based on tacrolimus, to prevent early rejection, and a double regimen to prevent late rejection.356,377
Immunosuppression and IBD post-liver transplantThe relationship between post-LT IS and IBD is bidirectional. IS can influence the course of IBD post-LT,378 while inflammatory activity from IBD can promote PSC recurrence.373 Patients diagnosed with IBD before LT experience more disease flare-ups and increased endoscopic and clinical activity post-LT. Additionally, new onset of IBD after LT is not uncommon.378
Regarding the effect of IS on IBD after LT, a systematic review including 14 studies described a deleterious effect of tacrolimus, contrasting with a favorable effect associated with the use of azathioprine379; similar conclusions were reached in a retrospective study by Mouchli et al.380 Montano-Loza et al.362 in a systematic review on the recurrence of liver immune-related diseases also reported an association between tacrolimus use and the appearance of de novo IBD post-LT. Despite this data, at present, with a wide array of biological drugs proven safe in the LT context, the potential differences or effects of post-LT IS are less relevant than in previous eras.381
The second part of the relationship between post-LT IS and IBD is the effect of uncontrolled intestinal inflammation on PSC recurrence in the graft. Published data indicates a relationship between IBD activity and PSC recurrence, both in post-LT follow-up and when LT is performed in the presence of active IBD.382 Thus, American and European guidelines recommend the use of 5-ASA before and after LT for inducing and maintaining remission in IBD associated with PSC. Thiopurines, especially azathioprine, can be used as maintenance drugs before and after LT, as they do not influence the incidence of postoperative problems or the risk of PSC-associated neoplasms.3,193 Regarding the use of biological drugs for IBD, ESOT emphasizes aspects related to safety, especially regarding the risk of opportunistic infections due to the combination of these products with the usual immunosuppression of LT. Specifically regarding anti-TNFα agents, caution is advised in patients with a history of bacterial cholangitis, and their concomitant use with antimetabolites following LT is generally discouraged.356
Biliary anastomosis in LT for PSCAnother controversial point until recent dates has been the need for biliary-enteric anastomosis in LT for PSC. These anastomoses preclude endoscopic access in case of need and increase the risk of ascending cholangitis. The indication for biliary-enteric anastomosis in LT for PSC was based on the potential inflammatory or neoplastic involvement of the recipient's biliary remnant. However, this type of anastomosis is associated with a higher incidence of ascending cholangitis and non-anastomotic strictures.383,384 Given the published evidence on choledochol-choledochal anastomosis, particularly concerning the risk of CCA in the residual bile duct, as well as the one-year incidence of anastomotic stricture or bile leakage, the current recommendation is that, whenever feasible, choledocho-choledochal anastomosis should be the preferred approach.3,356
What follow-up should be performed for a patient with PSC if she becomes pregnant?Recommendations:
- •
Close monitoring of patients is advised during and after pregnancy because of an increased risk of biochemical deterioration and pruritus. (LoE 4, strong recommendation).
- •
Liver ultrasound should be performed in patients with biochemical deterioration or pruritus onset. MRI is safe during pregnancy, although the use of a 1.5YT or lower MRI is suggested in the first trimester. (LoE 3, strong recommendation).
Pregnant women with PSC should be informed of the potential disease-related complications, particularly abdominal pain and pruritus.385 Although no standardized follow-up protocol during pregnancy has been universally established, it is considered reasonable to perform routine blood tests and abdominal ultrasound examinations once per trimester.386 Most patients remain clinically stable during pregnancy, particularly those receiving treatment with UDCA.387
In case of any clinical deterioration, such as worsening cholestasis or pruritus, abdominal ultrasound is warranted to exclude the presence of biliary strictures or other PSC-related complications. MRI is generally regarded as safe during pregnancy; however, it is recommended to limit imaging to 1.5YT or less during the first trimester. The use of gadolinium-based contrast agents is contraindicated unless absolutely necessary due to potential fetal risks.388
In the event of a dominant stricture in a symptomatic patient, therapeutic ERCP, including dilation and/or stent placement, can be safely performed when appropriate pregnancy-specific protocols are implemented.389
Does the presence of PSC affect pregnancy development, including fetal death, malformations, and preterm birth?Recommendation:
- •
Preconception counseling regarding the increased risk of complications during pregnancy should be provided to patients with PSC. (LoE 5, strong recommendation).
Pregnancy outcomes in women with PSC are influenced by several factors including the coexistence of IBD and the presence or absence of cirrhosis. Overall, pregnancies in patients with PSC are associated with an increased risk of adverse fetal outcomes. Reported live birth rates range from 80.6% to 100%, while preterm deliveries occur in approximately 7.7–25% of cases.390 The incidence of spontaneous miscarriage and stillbirth has been documented to range from 16% to 21.9%.387,390 Notably, UDCA treatment did not appear to significantly modify obstetric outcomes. Additionally, there is a higher likelihood of cesarean delivery among women with PSC (29.4%) than among the control population (13.3%).391 Despite these risks, fetal development and the incidence of congenital malformations generally remain within normal ranges.391
Primary sclerosing cholangitis in childrenIs the measurement of immunoglobulins and autoantibodies indicated for the diagnosis of autoimmune sclerosing cholangitis in pediatric patients?Recommendation:
- •
The evaluation of IgG and autoantibodies is required for the diagnosis of autoimmune liver diseases (AILD) in children. (LoE 4, strong recommendation).
- •
Determination of p-ANCA is advisable since ASC patients are more likely to be positive. (LoE 4, weak recommendation).
- •
In children autoantibodies titers should be considered positive in lower cut-off value than in adults. (LoE 4, strong recommendation).
The presence of autoantibodies is a cornerstone not only in the diagnosis of AILD but also in monitoring disease activity. In fact, serological markers are included in the diagnostic criteria for AIH established by the International Autoimmune Hepatitis Group,392 and modified for the pediatric age group by Mieli-Vergani et al.393
A high proportion of children with AILD test positive for autoantibodies (71–100%).394–396 ANA, SMA, or a combination of both are equally frequent, with similar titers in autoimmune sclerosing cholangitis (ASC) and AIH, while the presence of anti-LKM1 is significantly higher in AIH.394,395,397 For AILD diagnosis, low specificity has been described for ANA (65%), but very high specificity for SMA (95.5%), anti-F-actin (97.3%), anti-LKM1 (98.5%), anti-LC1 (99.3%), and anti-SLA/LP (100%).397 When comparing ASC to PSC, the presence of ANA and SMA is significantly higher in ASC.398,399 Anti-LKM1 is not present in PSC but is found in ASC.396,398–400 Despite this, the presence of autoantibodies is not essential for the diagnosis of AILD. Seronegative AIH is a well-recognized phenomenon in adults but is less described in pediatrics.401 It is worth noting that with current diagnostic scores, seronegative AIH is the most common cause of false negatives.402 On the other hand, seropositivity for p-ANCA is more frequent in ASC (74–80%) but can also occur in AIH (36–43%).395,398,399,411
In pediatric populations, autoantibody titers should be considered positive at lower threshold values compared to adults. While healthy adults may exhibit serum positivity at a 1/10 dilution without clinical significance, necessitating a cutoff point of 1/40, pediatric patients typically do not display autoantibody reactivity. Therefore, positivity at dilutions of 1/20 for ANA and SMA, and 1/10 for anti-LKM1, is deemed clinically significant in this demographic.403
What is the role of liver biopsy in children with PSC?Recommendations:
- •
Given the high incidence of ASC, liver biopsy should be performed in children with PSC. (LoE 4, strong recommendation).
- •
In cases of AIH with poor evolution and/or development of biliary features during follow-up, repeat liver biopsy is recommended to assess the possible progression to ASC. (LoE 4, strong recommendation).
- •
Liver biopsy should be considered as the gold standard for evaluating the severity of liver damage in pediatric patients. (LoE 4, strong recommendation).
There is ongoing debate regarding whether AIH and ASC (a term used to denote PSC/AIH in pediatric populations) constitute a spectrum of the same disease or if ASC represents a pediatric variant of PSC. The fact that almost half of the patients with AIH have features of sclerosing cholangitis in MRCP and the lack of differences in clinical presentation and survival between the two conditions suggests that they may be part of the same disease spectrum.404,405 Some authors have reported that up to 35% of children with PSC show features of AIH, suggesting that pediatric PSC has a more parenchymal and non-cholestatic presentation than adult PSC.406,407 Studies indicate that ASC associated with IBD and positive p-ANCA are linked to poor prognosis and low 10-year TFS, suggesting that these cases may represent pediatric variants of PSC.408
It is important to note that, in children, histological alterations are often nonspecific, requiring their interpretation alongside clinical, analytical, and radiological data. Indeed, Rojas et al. confirmed the similarities of histological features between AIH and ASC409 and MRCP is sometimes the only effective diagnostic method for their differentiation.410 For instance, periductal inflammation suggests the presence of variant syndrome, although it may be present in a small proportion of patients with AIH. Moreover, no histological alterations are detected in a significant percentage of ASC cases with abnormal MRCP. Conversely, nearly 30% of AIH patients may present biliary histological changes (bile duct damage, acute and/or chronic cholangitis, and biliary periportal hepatitis).404 Additionally, mild focal narrowing and contour irregularity without proximal dilation can be nonspecific changes of chronic liver disease.
For some authors, the role of liver biopsy in pediatric PSC is controversial, as histology provides limited information in the presence of MRCP abnormalities.411 However, other authors advocate for systematically performing liver biopsy due to the high rate of associated ASC (20–50%).404,405 In small duct sclerosing cholangitis, characteristic alterations are evident only on histology, despite a normal MRCP.412
Diagnostic criteria for AIH specifically designed for the pediatric population have been proposed by the IAIHG and histological abnormalities are part of these criteria. Therefore, liver biopsy is necessary to confirm the diagnosis and assess the severity of the disease. The 2008 simplified criteria for AIH demonstrated moderate sensitivity (77%) and high specificity (95%) for diagnosing AIH in children.413 For the appropriate application of these diagnostic criteria, it is essential to consider that AIH and PSC may occur simultaneously; or during the disease.414 Thus, in cases of AIH with poor evolution and/or development of biliary features during follow-up, repeat liver biopsy is recommended to evaluate the potential progression to ASC.
Liver histology remains the gold standard in children for assessing the severity of fibrosis, predicting survival, and identifying both overlap syndromes and other concomitant diseases.415,416
Is it necessary to rule out other autoimmune diseases in pediatric patients with sclerosing cholangitis? Which ones?Recommendation:
- •
In children with ASC, extrahepatic autoimmune diseases (EAD) should be excluded, not only at diagnosis, but also during follow-up. The search must be active, because it may be subtle and easily missed. (LoE 4, strong recommendation).
- •
In children with EAD and abnormal liver function tests of unknown cause, ASC should be examined. (LoE 4, strong recommendation).
Limited literature describes the association between AILD and extrahepatic autoimmune diseases (EAD) in pediatric populations. Few pediatric studies have shown a strong association between both entities, consistent with the findings of adults. EAD should be actively ruled out in children with AILD as symptoms can be subtle and underdiagnosed. Screening should be done at diagnosis and during follow-up, as EAD may develop before, during, and after AIH diagnosis. Additionally, AILD should be investigated in children with EAD showing abnormal liver function tests of unknown etiology.
The early diagnosis of EAD not only has important prognostic implications, such as the initiation of early IS therapy, but also supports the diagnosis of AILD. The IAIHG included EAD in its diagnostic scoring system,147 and was retained in the modified scoring system for the pediatric population by Mieli-Vergani et al.417
In this setting, IBD, mainly UC is the most associated extrahepatic disease,404,418,419 followed by autoimmune thyroid disease, celiac disease, autoimmune skin disease, and type 1 diabetes.404,418–422 Celiac disease and IBD are particularly significant because of the risk of IS treatment malabsorption, leading to insufficient response. Rare extrahepatic autoimmune conditions have been described in children with AIH type 2, including autoimmune polyglandular syndrome type 1 (APECED), 22q13 deletion syndrome, and infantile autoimmune hemolytic anemia with giant cell hepatitis.421
Paolella et al. found a strong association between AILD and EAD in a pediatric cohort (46%). UC was the most frequent (18.7%), followed by autoimmune thyroid disease (10%) and celiac disease (10%). In 8 of 9 cases with ASC, UC was detected, reinforcing the strong association between both conditions.418 Additionally, a systematic review and meta-analysis of nine studies involving 2046 pediatric patients detailed a pooled prevalence of celiac disease in children with AIH and vice versa of 6.3% (95% CI 3.87–11.7) and 1.4% (95% CI 0.84–2.15). Hepatic involvement in celiac disease can also include severe forms of AIH and, more rarely, ASC and PSC.420
Should screening for inflammatory bowel disease be performed in pediatric patients with PSC? When and how should it be conducted?Recommendation:
- •
Pediatric patients with sclerosing cholangitis should undergo fecal calprotectin testing at diagnosis to screen for IBD. The test should be repeated during follow-up if IBD-related symptoms arise. An elevated fecal calprotectin level warrants endoscopic evaluation. (LoE 4, strong recommendation).
The association between PSC-IBD in the pediatric population is well-established, with its prevalence reaching up to 90%.423,424 The onset is not always concurrent; IBD may precede liver disease for years, be diagnosed simultaneously, or emerge during follow-up.393,425 The phenotype of pediatric PSC-IBD has been less described than in adults, but most authors agree that it represents a milder liver disease phenotype with a more favorable prognosis,423,426–428 as it is associated with a lower frequency of cirrhosis and a lower rate of LT.423,424,428 In children with PSC-IBD, the intestinal phenotype is typically mild in severity and is significantly associated with pancolitis and rectal sparing.423,426,429,430 Age and gender do not appear to differ significantly between children with PSC-IBD and those with IBD alone.426,429,430
Several hypotheses propose that optimal management of intestinal disease may lead to a more favorable course of PSC. Its early identification could therefore have important prognostic implications in a condition that may progress to a fatal outcome. Thus, non-invasive screening for IBD should be considered in all patients who present with PSC and/or those who develop symptoms compatible with IBD during the disease. For this purpose, fecal calprotectin is the non-invasive marker of choice due to its good sensitivity and specificity. A meta-analysis published in 2017 highlighted excellent sensitivity and moderate specificity for detecting pediatric IBD, 97.8% and 68.2%, respectively.429 Subsequently, Ricciuto et al. demonstrated that fecal calprotectin correlated very well with endoscopic findings in pediatric patients with PSC-IBD, with an area under the receiver operating characteristic curve of 0.94 (optimal cutoff point, 93mg/g; sensitivity of 100% and specificity of 92%).431 This correlation was much superior to that provided by the symptom-based clinical activity index. Children with PSC-IBD in clinical remission, according to the symptom-based clinical activity index, have a significantly higher risk of active endoscopic and histological disease compared to children with IBD without PSC, although clinical indices may underestimate disease severity.431,432 Endoscopic examination should be reserved for cases with elevated fecal calprotectin and/or those associated with clear symptoms.
How is the prognosis of PSC evaluated in pediatric patients?Recommendations:
- •
Caution is recommended when applying diagnostic criteria and prognostic scores developed for adults to pediatric patients with PSC, given the significant clinical, biochemical, and histological differences between these populations. (LoE 4, strong recommendation).
- •
The SCOPE Index is the sole risk stratification and prognostic tool that has been specifically validated for use in pediatric patients with PSC, and it is currently the only tool suggested for this population. (Statement, consensus 100%).
As mentioned before, several predictive models have been developed using data from adult-onset PSC populations. However, there is no consensus on the optimal model to predict prognosis in pediatric population433–435 as none of them has been validated in children.436
There are notable clinical differences between pediatric and adult-onset PSC: (1) at the time of PSC diagnosis, dominant strictures are present in 4–16%437,438 of children, which is significantly lower than the figures reported in adults,439 (2) CCA is rare in pediatric onset PSC, occurring in only 1% of children within 10 years,397,438 (3) the small duct phenotype much more common in children (20% of children with PSC),437,438 (4) features of AIH overlapping with PSC are observed in over 30% of children,437,438 (5) due to bone turnover in growing children, ALP is not a reliable indicator of liver disease in this population, and (6) other risk factors for liver disease progression, such as chronic alcohol abuse, smoking, obesity, and polypharmacy, are less prevalent in children.436
Given these disparities, it remains uncertain whether risk models derived from adult patients can be applied or generalized to pediatric cases.436 Although children with PSC are susceptible to developing cirrhosis and end-stage liver disease, with approximately 30% requiring LT within 10 years of diagnosis, there are currently no widely accepted prognostic scores for use in pediatric patients with PSC.
The Sclerosing Cholangitis Outcomes in Pediatrics (SCOPE) index, published in 2021 using data from the Pediatric PSC Consortium, is the only pediatric-specific risk stratification and prognostic tool validated for use in pediatric patients. This index includes various parameters, including bilirubin, albumin, GGT, platelet count, and findings of multifocal large duct beading and strictures on MRCP or ERCP, to assess a patient's risk. Initially, the first retrospective group of patients consisting of 1012 patients from 40 centers was used to create a multivariate risk index. The model classifies patients into low-, medium-, and high-risk groups based on their annual rates of progression to transplant or death. The combined rate of LT or liver disease-related death varies among these risk groups, less than 1% per year for low-risk patients, approximately 3% and 9% per year for medium- and high-risk patients, respectively. Furthermore, the SCOPE index estimates the likelihood of hepatobiliary complications and CCA. The rates of progression to hepatobiliary complications were <2%, 6%, and 13% for the low-, medium-, and high-risk groups, respectively. External validation of the model in a cohort of 240 children across 11 independent centers confirmed its satisfactory performance.440
Recently, non-invasive quantitative MRI metrics, including MRI-derived liver and spleen stiffness, have shown considerable promise as biomarkers for the detection of AILD and the monitoring of disease progression in children and young adults.441–443 However, only a few studies have assessed the relationship between MRI-based markers of liver disease and clinical risk scores. In a prospective study, 58 patients diagnosed with AILD, including 16 with PSC, underwent liver MRI examinations incorporating MR elastography. The results revealed that increased liver stiffness and larger bile duct diameters were independently associated with higher Mayo risk scores435 and SCOPE index440 among children and young adults with AILD. Although the study focused on pediatric cases, it is essential to consider limitations such as the small number of reported cases and the heterogeneity of the sample, including patients with AIH and PSC. Moreover, it is important to acknowledge that MR elastography is not universally available and remains inaccessible in many centers.
Previous studies have shown that LSM using VCTE correlates independently with the histological fibrosis stage. Cut-off values of 9.6kPa for extensive fibrosis and 14.4kPa for cirrhosis yield diagnostic accuracies exceeding 0.80.444 However, in the pediatric population, there are no data validating the use of LSM to assess fibrosis status in patients with PSC or to correlate it with clinical events.
Is there a prognostic value in the decrease in GGT value or its normalization in pediatric patients with sclerosing cholangitis?Recommendation:
- •
Normal GGT levels at diagnosis and a decrease in GGT levels during disease progression (irrespective of treatment status) can be associated with favorable outcomes in pediatric PSC patients. (Statement, consensus 100%).
In children, ALP levels tend to fluctuate widely due to bone growth; therefore, GGT is commonly used as a marker for cholestasis in pediatric patients.445 Moreover, higher APRI, GGT, and bilirubin values are generally associated with worse long-term outcomes.437 Patients with even mild elevation of these markers at baseline usually have more advanced disease and are likely to experience worse outcomes. However, GGT levels fluctuate widely and often, especially in early stages of the disease in children with PSC. Therefore, individual GGT levels should be considered cautiously.446
In a retrospective study involving 287 pediatric patients with PSC, it was observed that those with GGT levels<50YIU/L one year after diagnosis exhibited a lower likelihood of experiencing hepatobiliary complications (PH, biliary complications, CCA, LT, or death from liver disease). The reported 5-year event-free survival rate in the GGT<50UI/L group was 91% compared with 67% in patients with GGT levels≥50YIU/L after one year.
The extent of GGT reduction from diagnosis to one year also correlates with 5-year event-free survival, with rates of 88%, 78%, and 61% observed when reductions were greater than or equal to 75%, between 26% and 74%, and less than or equal to 25%, respectively.447 In another multicenter retrospective review conducted using data from the PSC Consortium, 263 pediatric patients with GGT levels>50YIU/L at diagnosis and undergoing UDCA treatment for at least one year were analyzed. Normalization of GGT at 1 year was achieved in 46% of patients and was associated with a more favorable 5-year outcome, with native liver survival reaching 99% in those who achieved GGT normalization, compared to 77% in those who did not.448
These findings are consistent with those of a single-center cohort study, which observed that failure to achieve at least a 25% reduction in GGT within the first year following a diagnosis of pediatric-onset PSC was strongly associated with the early occurrence of adverse clinical events.446
Does pediatric PSC-IBD phenotype have a better prognosis?Recommendations:
- •
Pediatric patients with PSC-IBD exhibit a slower progression of liver disease than those without IBD. (Statement, consensus 100%).
- •
It is recommended to consider the increased risk of CRC, CCA, and post-transplant disease recurrence when evaluating prognosis and management strategies in patients with PSC-IBD. (LoE 4, strong recommendation).
Between 70% and 80% of pediatric patients with PSC concurrently present with IBD. Among these cases, above 80% are diagnosed with UC or indeterminate colitis, while 15–20% are diagnosed with Crohn's disease.437,438,449 IBD is typically diagnosed within three months of PSC diagnosis or even earlier, with less than 10% of cases diagnosed with PSC preceding IBD by more than three months.438,449 The interplay between PSC and IBD may define a less severe patient phenotype that could impact the course of both conditions. On one hand, there have been two studies suggesting that PSC may offer protection against the need for colectomy due to chronically active or medically refractory colitis.450–452 Several authors have suggested that pediatric-onset PSC also runs a milder course and has a more favorable outcome than adult-onset PSC, especially when it is associated with IBD.437,438 According to the PSC Consortium data, patients with PSC-IBD had a lower rate of cirrhosis and higher survival rate with the native liver. The total proportion of patients with end-stage liver disease and PH at baseline was similar between PSC and PSC–IBD patients, however PSC–IBD patients had a slower progression to complications.437 Despite this slow progression, the impact of this supposedly milder phenotypic presentation on the need for transplantation remains debatable. In a large longitudinal cohort study published by Valentino et al., the rate of cirrhosis was not significantly different between patients with PSC and concomitant IBD compared to those without (22% vs 41%, p=0.06), but the former group had a lower rate of LT (2% vs 18%, p=0.01).438 However, among LT recipients for PSC documented in the SPLIT (registry gathering data on children under 18 years of age who underwent LT across 45 pediatric transplant centers in the United States and Canada since 1985) and UNOS databases, approximately 60% were diagnosed with IBD at some stage of their disease progression.453,454 Moreover, a negative correlation has been observed regarding the rate of post-transplant PSC recurrence among patients included in the SPLIT Registry and data from the Pediatric PSC Consortium.453,455 In another single-center study reporting their experience with LT in patients with PSC, all cases of recurrent PSC occurred in individuals with concomitant IBD.455
Other risk factors that could influence outcomes have also been identified.450 In a study of 9405 patients with childhood-onset IBD, patients with PSC-IBD were at a greater risk of developing cancer than those in the general population.456 In a prospective case series from 20 European countries and Israel studying 60 patients with childhood-onset IBD who developed a malignancy or died, 56% of patients with CCA or CRC were also diagnosed with PSC.457 In a systematic review conducted by Olen et al., involving 9442 individuals with pediatric IBD, 294 deaths were reported. Excluding patients with very early-onset IBD, the highest relative risk was observed in those with UC and concomitant PSC (HR, 12.2; 95% CI, 8.4–17.8).458
What is the role of UDCA in the treatment of PSC in children?Recommendations:
- •
Treatment with UDCA at standard doses (15mg/kg/day) can be considered for pediatric patients with PSC, as it may be associated with higher rates of GGT normalization and improved outcomes, including increased TFS and fewer hepatobiliary events. (LoE 4, weak recommendation).
The role of UDCA in modifying the disease course and improving survival of patients with PSC remains controversial. Due to the limited data available in the pediatric population, formal recommendations are lacking. Nevertheless, UDCA continues to be used in at least 80% of pediatric PSC cases.437
As previously noted, patients who achieved GGT normalization, defined as a level<50YIU/L within one year or a reduction of ≥75% from baseline, exhibited improved survival compared to those who did not. Notably, many untreated children also achieve spontaneous GGT normalization following diagnosis.8 While the survival benefit remained consistent irrespective of whether patients achieved GGT normalization through UDCA treatment or spontaneously without intervention, the incidence of GGT normalization was notably higher among patients receiving UDCA treatment compared to those who did not (53% vs 29%, respectively). Consequently, the percentage of patients with favorable outcomes was higher in those treated with UDCA.447 Moreover, in a retrospective study of pediatric PSC patients treated with UDCA for at least one year, the 5-year survival rate with native liver was 99% in patients who achieved GGT normalization vs. 77% in those who did not.448 Controlled withdrawal of UDCA from children receiving chronic therapy with normalized liver biochemistry demonstrated that most children did not experience biochemical flares.459 One-third of the patients in the study maintained a completely normal GGT level, raising the question of whether they derived any therapeutic benefit from UDCA or whether their biochemical normalization would have occurred spontaneously.
In a retrospective study utilizing data from the Pediatric PSC Consortium, children administered oral vancomycin were matched in a 1:1:1 ratio with those treated with UDCA and those receiving no treatment. The findings indicated that neither oral vancomycin nor UDCA resulted in improved outcomes compared to an observational approach, as patients across all treatment groups exhibited similar rates of progression to end-stage liver disease. Notably, spontaneous normalization of biochemistry was frequently observed in untreated children, particularly among those with a mild phenotype and early-stage disease.460
What is the role of LT in PSC in pediatric patients?Recommendations:
- •
LT should be considered in patients with PSC who develop complications such as PH, end-stage liver disease, or CCA. However, the risk of disease recurrence should be considered when evaluating post-transplant prognosis. (LoE 4, strong recommendation).
- •
PSC-IBD is associated with an increased risk of recurrence after LT. (Statement, consensus 100%).
In a population-based natural history registry of pediatric patients diagnosed with PSC, LT was indicated in 14% of the cohort.437 This occurred at a median age of 15 years, approximately four years after the initial diagnosis of the liver disease. Patient survival was 88% and 70% at 5 and 10 years, respectively.437,438 Additional single-center studies reported significantly higher transplantation rates. However, these findings should be interpreted with caution, as they are often derived from LT centers that predominantly receive patients referred for transplantation.461,462
Considering the data from this registry, approximately 3% of patients received a LT for PSC.463 Children listed and transplanted for PSC were significantly older than those transplanted for other indications. While cirrhosis complications and end-stage liver disease remain the primary indications for LT, some cases of LT for CCA have been reported. The identification of PH or biliary complications represents a tipping point in the natural history of PSC, with a subsequent median survival of only 3 years with a native liver.437
Data from both the UNOS and SPLIT registries indicate that early post-transplant survival in children with PSC is higher than in those transplanted for other indications.453,454 According to data from the SPLIT registry, 10% of pediatric patients transplanted for PSC experienced disease recurrence at a mean of 18 months following the initial transplant (mean follow-up duration: 3 years). However, the recurrence rate may be increased with an extended follow-up. IBD is associated with an increased risk of mortality and recurrent PSC. No statistically significant differences in immunosuppressive regimens have been identified between patients who developed recurrent PSC and those who did not.453 According to data from the Pediatric PSC Consortium, recurrence rates of PSC after LT are 10% at 2 years and 27% at 5 years post-LT. Patients who developed recurrent PSC underwent transplantation at a younger age (12.9 vs. 16.2 years) and had a shorter interval from PSC diagnosis to LT (2.5 vs. 4.1 years), and higher ALT at the time of transplantation (112 vs. 66YIU/L). IBD prevalence was higher in the recurrent PSC group (86% vs. 66%). After LT, patients with recurrent PSC experienced a higher number of biopsy-proven acute rejection episodes and had a greater incidence of steroid-refractory rejection. Additionally, 43% of those with recurrent PSC developed PH, were re-listed for LT, or died within two years of diagnosis. Mortality was significantly higher in the recurrent PSC group (11.1% vs. 2.9%).455
Is MRCP indicated in pediatric patients with AIH?Recommendations:
- •
MRCP should be performed in children diagnosed with AIH to evaluate bile duct abnormalities that are compatible with ASC. (LoE 4, strong recommendation).
- •
If clinical or analytical parameters of cholestasis are established during AIH follow-up, MRCP should be repeated to rule out signs of ASC. (LoE 4, strong recommendation).
- •
Differential diagnosis between AIH, PSC, and ASC can be complex because of their similarities, and current diagnostic scoring systems do not allow for clear differentiation. Therefore, in addition to clinical manifestations, biochemical and serological alterations, and histopathology, imaging techniques must be used to distinguish between these conditions.441 International clinical practice guidelines recommend performing cholangiographic studies in all children with AIH to rule out stenosis and biliary dilatations compatible with ASC.412,417,464 Since the two diseases are not always present simultaneously, the emergence of clinical or analytical signs of cholestasis during the follow-up of AIH should be repeated to exclude ASC.
- •
MRCP is the imaging diagnostic method of choice in children with autoimmune liver disease, both at presentation and during follow-up, as it exhibits good sensitivity (81–84%) and specificity (94–100%) for the diagnosis of pediatric PSC.465,466 However, as a qualitative technique, it is subject to inter-observer variability. More recently, quantitative MRCP (MRCP+) has emerged as a novel imaging processing tool that provides quantitative metrics derived from 3D MRCP images obtained in a relatively short time.467 It allows for objective assessment and measurement of the volume, length, and diameter of the biliary tree, thereby highlighting the differences between ASC and AIH. Additionally, it provides information on the degree of inflammation and fibrosis in the hepatic parenchyma. Gilligan et al. asserted that MRCP+ had an excellent capacity to differentiate between AIH and ASC in children and monitor their follow-up.441 Recently, a prospective study with a cohort of 66 children described good diagnostic predictive capacity for MRCP+ (AUC 0.91) when combining the number of dilatations with the relative severity of strictures and dilatations.443 In this regard, other authors have demonstrated the use of MRCP+ as a prognostic marker in children with sclerosing cholangitis.468–470 Patil et al. proposed modifications to the Majoie classification for ERCP and concluded that an MRCP-based scoring system incorporating the most affected intrahepatic and extrahepatic regions was both reliable and predictive of ASC progression in pediatric patients.468 Mahalingam et al. also incorporated the prognostic capability of MRCP+ in children and young adults with AILD, observing associations with serum fibrosis markers and other imaging techniques (magnetic resonance elastography-derived liver stiffness, T1 relaxation measurements).470
Recommendations:
- •
Early initiation of standard AIH treatment is recommended in patients with ASC, as it may positively influence the progression of parenchymal involvement. (LoE 4, strong recommendation).
- •
Immunomodulatory therapy is not recommended in patients with isolated PSC, as it has not been associated with improved outcomes. (LoE 2, strong recommendation).
Some authors have suggested that early initiation of treatment may lead to favorable responses to parenchymal liver damage in ASC. This response includes normalization of biochemical and immunological parameters with the same immunosuppressive treatment utilized for AIH, resulting in good medium-to long-term survival outcomes. However, the bile duct disease progresses in about 50% of patients despite treatment,404 particularly in those with associated difficult-to-treat IBD. In a retrospective study comparing treatment response and outcomes in children with ASC, no significant differences were observed between the two groups, both demonstrating a favorable response to prednisolone with or without azathioprine.405 However, unlike the initial study, the authors diagnosed ASC only in patients who developed cholestatic manifestations during follow-up without performing cholangiographic evaluation at the time of presentation. This difference precludes direct comparison between the two studies.
Most other published series on pediatric sclerosing cholangitis are retrospective single-center studies with small patient cohorts. In all retrospective studies, a variable proportion of patients present with variant syndrome, however, while some reports describe a favorable response to immunosuppressive therapy and a better prognosis compared to PSC,406,462,471,472 in others the prognosis of PSC-AIH is reported to be severe and not ameliorated by immunosuppressive treatment473 or similar to that of PSC irrespective of treatment.437,461,474
In a series published by Valentino et al., out of 120 patients diagnosed with PSC, 31 had features of AIH. A higher proportion of patients with ASC were receiving corticosteroids and immunomodulators than those with isolated PSC. However, it cannot be concluded that this difference was associated with improved biochemical outcomes, as ASC patients presented significantly higher ALT levels at 1 year than those with isolated PSC.45,120 A potential difference may lie in the need for LT: of the six patients who underwent transplantation in this cohort, five had isolated PSC and only one had variant syndrome.
Other studies have demonstrated that corticosteroids, azathioprine, methotrexate, and infliximab do not appear to alter PSC progression. The prevalence of AIH variant is notably higher in patients without IBD. It is possible that concurrent immunosuppressive therapy for IBD may have masked PH–driven inflammation characteristic of AIH in some patients, leading to misclassification as isolated PSC cases.404,475,476
Genetic cholestasisWhat genetic alterations are present in PFIC1? What is the function of this mutated gene? Is it necessary to conduct family segregation studies? Is there a genotype–phenotype correlation in PFIC due to FIC1 deficiency?- •
Genetic investigations should be performed on first-grade relatives of the index case. (LoE2, strong recommendation).
The ATP8B1 gene, located on chromosome 18q21–22, encodes the FIC1 protein, a P-type ATPase flippase involved in the translocation of phospholipids from the outer to the inner leaflet of the hepatocanalicular membrane bilayer.477–480 Although the precise pathophysiological mechanisms of cholestasis in FIC1 deficiency are not fully understood, it is believed that the loss of canalicular phospholipid asymmetry compromises the structural integrity of the membrane, rendering it more susceptible to detergent effects of hydrophobic bile salts. This disruption may also impair the function of key transmembrane transporters involved in bile formation, such as the bile salt export pump (BSEP).481–483 Moreover, FIC1 is expressed in the canalicular membrane of hepatocytes and cholangiocytes,477 and its presence is notable in extrahepatic tissues such as the small intestine,479 pancreas, kidney, lung, and cochlear cells. This widespread distribution accounts for the extrahepatic manifestations observed in cases of severe FIC1 deficiency.25 A down-regulation of FXR, a nuclear receptor of BA, has been proposed as an additional mechanism contributing to cholestasis in FIC1 deficiency.484–486 FXR downregulation causes secondary inhibition of BSEP and increased expression of the ileal apical sodium-dependent bile acid transporter (ASBT), leading to bile acid overload.
No genotype–phenotype correlation could be established in PFIC1 in a retrospective multicenter international series from the NAPPED Consortium.487,488 Pathogenic biallelic mutations in ATP8B1 are associated with a spectrum of clinical presentations489 ranging from the less severe, intermittent form of the disease, formerly known as benign recurrent intrahepatic cholestasis type 1 (BRIC1), to the more severe and progressive condition termed PFIC1.490–494 In addition, heterozygous pathogenic variants of ATP8B1 have been implicated in transient neonatal cholestasis,495 ICP496 and some instances of BRIC1.491 It is important to acknowledge that following the identification of an index case with such pathogenic mutations, it is recommended to carry out genetic analysis on the parents and siblings to elucidate the genetic landscape and enable accurate genetic counseling.
Which genetic alterations are present in PFIC2 (BSEP)? What is the function of the mutated gene? Is it necessary to conduct family segregation studies? Is there a genotype–phenotype correlation in PFIC due to BSEP deficiency?Recommendations:
- •
Genetic analysis should be performed on the parents and siblings after the diagnosis of the index case. (LoE 2, strong recommendation).
ABCB11, which is located on chromosome 2q24, encodes the BSEP protein, an ATP-binding cassette (ABC) transporter.497,498 BSEP is specifically expressed in the canalicular membrane of hepatocytes, where it functions as a bile salt export pump, facilitating the excretion of bile salts from hepatocytes into the bile canaliculus.498 Mutations in ABCB11, which are biallelic and pathogenic, have been shown to cause a spectrum of liver diseases.489,497 The severity of these conditions can range from less serious and intermittent, formerly known as BRIC2, to more severe and progressive type PFIC2.499 There have been reported cases in which patients initially present with a phenotype resembling BRIC2, which then evolves into PFIC2.500 In cases of PFIC2, a genotype–phenotype correlation has been identified.501,502 Mutations in ABCB11, particularly splice and frame-shift mutations that result in either an absent or non-functional protein, are associated with a poorer prognosis than mutations that maintain some residual function. Specifically, missense mutations such as p.D482G and E.297G are associated with a more favorable outcome. Heterozygous mutations in ABCB11 have been associated with various liver conditions including transient neonatal cholestasis,489,503–505 ICP,506,507 drug-induced liver injury (DILI),506 and certain cases of BRIC2.21 Interestingly, common variants such as single nucleotide polymorphisms (SNPs) may play a role in the etiology of cholestatic phenotypes. For instance, the BSEP SNP p.V444A has been identified as a predisposing factor for ICP and DILI.506
Upon the diagnosis of an index case, it is recommended to perform genetic evaluation of both parents and siblings to ascertain the possible inheritance patterns and risks associated with ABCB11 mutations.
How can we diagnose defects in PFIC1 and PFIC2 in children?Recommendations:
- •
Confirmation of the diagnosis of PFIC1 and PFIC2 must be performed using genetic analysis. (LoE 2, strong recommendation).
- •
Performing a liver biopsy is not mandatory for the diagnosis, although it may be useful. (LoE 4, strong recommendation).
- •
The absence of BSEP canalicular staining supports the diagnosis of BSEP deficiency. Normal BSEP staining should not exclude BSEP diagnosis, as some ABCB11 mutations can affect the functionality of the protein but preserve normal synthesis. (LoE 4, strong recommendation).
PFIC types 1 and 2 are clinically overlapping yet genetically distinct disorders characterized by early onset cholestasis among other clinical manifestations.500,508 The definitive diagnosis of both PFIC1 and PFIC2 relies on genetic analyses. Pathological mutations, either compound heterozygous or homozygous in nature, must be identified within specific loci: ATP81B on chromosome 18 q21 for PFIC1 and ABCB11 on chromosome 2q24 for PFIC2. These genetic markers are crucial for confirming the PFIC type.
A thorough patient history is paramount in guiding the diagnostic process. Key elements, such as familial consanguinity,489,500 history of intrahepatic cholestasis of pregnancy, or established cases of PFIC within the family, can provide significant diagnostic insights.
The clinical presentations of PFIC1 and PFIC2 share numerous features, notably severe defects in FIC1 and BSEP function, respectively, resulting in persistent cholestasis in infancy, with onset during the first months of life.500,508 This form of cholestasis is characterized by normal GGT levels in relation to age, elevated serum bile acid concentrations, and the onset of severe pruritus, typically beginning at approximately 3 months of age.500 The presence of pruritus500,508 is a hallmark symptom and is often disproportionate to the degree of bilirubin elevation.18 While a later presentation or episodic jaundice may be indicative of PFIC, persistent jaundice is more characteristic of these conditions.500 Consequences such as growth retardation and significant deficits in fat-soluble vitamins500 are common to both PFIC1 and PFIC2. In PFIC1, a prototypical multisystem disorder, diarrhea500,508 may be the initial clinical manifestation.
However, differences in the clinical features can assist in distinguishing between PFIC1 and PFIC2. BSEP deficiency,500,508 the more prevalent of the two, is often accompanied by elevated alpha-fetoprotein levels and markedly higher aminotransferase values (ALT exceeding 5 times the upper limit of normal) as opposed to FIC1 deficiency (usually with ALT≤100YIU/L). Notably, FIC1 deficiency is associated with extrahepatic manifestations500,508 including recurrent diarrhea, pancreatitis, and sensorineural hearing loss, which are not observed in BSEP deficiency. The degree of growth impairment is notably more pronounced in PFIC1.500,508 Furthermore, BSEP deficiency increases the risk of HCC,489,500,508 a complication that has not yet been reported in FIC1.
Liver biopsy, which is not required for diagnosis, can yield auxiliary information. Although generally lacking specific diagnostic features, the presence of giant cell transformation or multinucleate hepatocytes is frequently observed in cases of BSEP deficiency.500,508
BSEP immunostaining of the liver tissue may be of diagnostic utility,500,509,510497,511 particularly when genetic analysis results are anticipated to be delayed. While in PFIC1, canalicular expression of BSEP remains normal, the absence of canalicular BSEP staining reinforces the diagnosis of BSEP deficiency. However, it is important to note that normal BSEP immunostaining does not rule out BSEP deficiency, as certain ABCB11 mutations may impair protein functionality while preserving normal synthesis and localization.
What is the typical clinical course of PFIC1 in children, and how should disease progression be monitored?Recommendation:
- •
Regular assessment of pruritus, PH, and hepatic insufficiency should be performed, as they may indicate the need for LT. (LoE4, strong recommendation).
- •
Patients must be referred to a specialized pediatric liver center. (LoE5, strong recommendation).
- •
Infants should receive formulas containing large amounts of medium-chain triglycerides (MCT). (LoE4 Strong recommendation).
- •
Fat-soluble vitamin levels must be monitored regularly. Early supplementation with vitamins A, D, E, and K must be initiated. (LoE4 Strong recommendation).
Systematic monitoring of the progression of PH and hepatic insufficiency is imperative in the management of PFIC. Patients with PFIC1 tend to experience more gradual progression to end-stage liver disease than those with BSEP deficiency. LT in PFIC1 is often indicated primarily because of intractable pruritus, which severely impairs the quality of life, rather than because of liver failure itself.489,500,508 At the age of 18 years, the probability of survival with the native liver is approximately 44%.487
It is essential that patients with PFIC be managed in specialized pediatric liver centers to ensure access to comprehensive and expert care. Nutritional evaluation is a critical component of this care, especially given that diarrhea, a common symptom of PFIC1, can complicate nutritional management. Nutritional interventions should address the increased energy demands of these patients15 by incorporating formulas enriched with MCT for infants to facilitate absorption and caloric intake.12,14 Monitoring fat-soluble vitamin levels is another essential aspect of PFIC management, aiming at preventing the development of deficiencies.512,513 Prophylactic supplementation with vitamins A, D, E, and K should be initiated early and adjusted as necessary based on regular monitoring.12,512,513
Routine assessment of pruritus and its impact on daily activities, sleep, and overall quality of life for patients and their families is recommended.18 Pruritus is not only a critical symptom in PFIC, but also a pivotal factor in therapeutic decision-making, potentially serving as an indication for LT when it proves refractory to medical management.
What is the typical clinical course of PFIC2 in children, and how should disease progression be monitored?Recommendations:
- •
Patients with PFIC2 should be referred to a specialized pediatric liver center. (LoE5, strong recommendation).
- •
Nutritional assessment is mandatory. Infants should receive formulas containing large amounts of MCT. (LoE4, strong recommendation).
- •
Prothrombin time and fat-soluble vitamin levels should be monitored regularly. Early supplementation with vitamin A, E, D, K should be started. (LoE4, strong recommendation).
- •
Frequent assessments of pruritus, liver function, and PH are strongly recommended. (LoE4, strong recommendation).
- •
Regular surveillance for HCC must be initiated upon the diagnosis of the disease. (LoE4, strong recommendation).
In the context of BSEP deficiency prognosis, recent findings indicate that native liver survival at the age of 18 is approximately 32%.487 It is imperative to consider the genotype of patients with BSEP deficiency, because a demonstrable correlation exists between genotype and prognosis.487,489,508,514 Specifically, patients harboring mutations that permit residual BSEP protein functionality, such as missense mutations p.D.482G and p.E297G,508 exhibit higher native liver survival rates.487 However, a recent analysis has shown that heterozygous patients bearing p.D482G or p.E297G mutations combined with a predicted protein-truncating mutation (PPTM, mutations leading to a complete absence of functional BSEP activity) have a similar prognosis to patients bearing two PPTM, with an expedited progression to hepatic insufficiency and subsequent PH, worse native liver survival, and poorer response to surgical biliary diversion.515 Furthermore, it has been observed that patients with severe BSEP deficiency progress to end-stage liver disease more rapidly than those with FIC1 deficiency, with earlier development of hepatic insufficiency and PH.489,500,508 Therefore, prompt referral of these patients to centers specializing in pediatric liver care is essential.
Nutritional assessment and intervention15 are of paramount importance in the management of patients with BSEP deficiency. Owing to increased energy demands and malabsorption issues, nutritional support is required, and infants should be provided with formulas enriched with higher quantities of MCT.12 Moreover, regular monitoring of fat-soluble vitamin levels is of critical importance,53,58 along with close surveillance of prothrombin time, to prevent complications related to vitamin deficiency. Proactive initiation of supplementation12,512,513 with vitamins A, E, D, and K is recommended. Additionally, frequent evaluations to assess hepatic insufficiency and PH are strongly recommended.
Pruritus is a significant symptom of BSEP deficiency, necessitating regular assessments to determine its impact on daily activities, sleep, and overall quality of life for both the patient and their family. The severity of pruritus is a crucial factor in therapeutic decision-making processes.
Finally, routine surveillance for HCC is advocated from an early age, given the potential risk in this population.487,489,500
What is the impact of genotype on the clinical course of PFIC associated with BSEP deficiency? Do mutations such as p.D482G or p.E297G confer a more favorable prognosis? What is the expected prognosis in cases with mutations leading to non-functional or truncated BSEP protein?- •
Genotype has a significant impact on the clinical evolution and prognosis of PFIC2. (Statement, consensus 100%).
Truncating, nonsense, or splice-site ABCB11 mutations, leading to a predicted absent or nonfunctional protein, are correlated with a more severe clinical outcome. The absence of functional protein expression results in a markedly reduced median survival of native liver, with reports indicating a median of only 3.5 years.487 Conversely, specific missense mutations such as p.D482G and E.297G allow for residual function of the BSEP and are associated with a more favorable prognosis,487 except when combined with a PPTM, as has recently been shown.515 Overall, the native liver survival (NLS) rate at 10 years of age was comparable between patients with two PPTM mutations (NLS 23%) and those with compound heterozygosity for either the p.D482G or p.E297G mutation, along with one PPTM mutation (NLS 21%). This rate was significantly lower than that observed in patients homozygous for either the p.D482G or p.E297G mutation (NLS 75%).515 Similarly, the response to surgical biliary diversion was poor in the first two groups compared to those homozygous for p.D482G or p.E297G, potentially identifying a group of patients who will likely not respond to surgical biliary diversion or IBAT inhibitor treatment.515 Therefore, one protein truncating mutation in ABCB11 would override the favorable phenotypic effect of a concomitant p.E297G or p.D482G mutations in ABCB11.515
Homozygous p.E287G or p.D482G mutations are associated with long-term native liver survival.515 Patients homozygous for p.D482G had less severe evolution than those homozygous for p.E297G. However, patients bearing one PPTM mutation and one p.D482G mutation had a more severe phenotype than those bearing one PPTM mutation and one p.E297G.515,510,548
Furthermore, there is a distinct correlation between genotype severity and HCC incidence in patients with PFIC2. Patients with biallelically predicted protein truncation mutations, which result in the complete absence of functional BSEP activity, exhibit a higher incidence of HCC.487,515
Which therapeutic alternatives are available for patients with PFIC1 and 2?Recommendations:
- •
LT should be considered for end-stage liver disease caused by PFIC1 or PFIC2. (LoE3, strong recommendation).
- •
IBAT inhibitors should be considered the first step in the treatment of moderate/severe pruritus. (LoE2, strong recommendation).
- •
Surgical biliary diversion (SBD) can be considered a non-pharmacological alternative before the development of advanced fibrosis or cirrhosis. (LoE4, weak recommendation).
- •
Monitoring BA is recommended as it is correlated with pruritus response and outcomes. (LoE2, strong recommendation).
LT remains a definitive therapeutic intervention for patients with end-stage liver disease resulting from PFIC1 or PFIC2. However, the management of pruritus has evolved considerably. Historically, treatment options15,22,515 were limited and predominantly consisted of off-label use of agents such as UDCA,516 phenobarbital, rifampicin, cholestyramine, and antihistamines. However, these interventions often fail to achieve satisfactory outcomes.
Advancements have focused on disrupting enterohepatic circulation to mitigate pruritus, primarily through SBD487,517–521 or the administration of IBAT inhibitors.522–524 While the efficacy of these approaches varies and not all patients respond favorably, IBAT inhibitors are increasingly considered the first-line therapeutic strategy for severe pruritus in PFIC populations, potentially reducing the need for SBD.
The current consensus suggests that SBD should be implemented before the development of advanced fibrosis or cirrhosis.525 In the context of BSEP deficiency, the nature of the mutation significantly influences therapeutic outcome.487,515 Severe mutations that result in nonfunctional proteins (PPTM mutations) render SBD ineffective. Therefore, it is not recommended in these cases. Conversely, patients with at least one favorable mutation, such as p.D482G or p.E297G in BSEP, have a considerably higher likelihood of achieving partial or complete pruritus remission following SBD.487 However, recent evidence indicates that heterozygous patients carrying a p.D482G or p.E297G mutation combined with a PPTM have a poor response to SBD, similar to that of patients with two PPTM mutations.515 Approximately 50% of patients with PFIC1 can anticipate a long-term response to pruritus following SBD.487 Notably, the total serum BA concentration or the rate of reduction in BA post-SBD correlates with the response to pruritus treatment and long-term prognostic outcomes.487
In patients receiving IBAT inhibitors, is a reduction in BA levels associated with improved native liver survival?Recommendations:
- •
BA monitoring is recommended as its evolution is correlated with native liver survival, growth, and quality of life in patients treated with IBAT inhibitors. (LoE3, strong recommendation).
Odevixibat and maralixibat are IBAT inhibitors approved for the treatment of cholestatic pruritus in children with PFIC. Maralixibat is approved for children from 3 months of age, and odevixibat from 6 months onward. These agents act by blocking the reuptake of BA in the terminal ileum, thereby promoting their fecal excretion and reducing systemic bile acid burden, while exhibiting minimal systemic absorption. Through this mechanism, they pharmacologically mimic the effects of surgical biliary diversion. IBAT inhibitors are generally well tolerated, with diarrhea and elevated ALT and bilirubin levels being the most frequently reported adverse effects.
Clinical data from the PEDFIC1 study522 showed that in children with PFIC1 and PFIC2, odevixibat significantly improved pruritus and reduced serum bile acid levels compared to placebo, with effects that were both rapid (in the first 4 weeks) and sustained over time. Improvements in the sleep parameters were also observed. Notably, patients with decompensated liver disease and those with BSEP deficiencies caused by two mutations that predict the absence of functional proteins were not included in this study.
Further evidence suggests that reduction in serum BA levels after 6 months of IBAT inhibitor therapy may serve as a prognostic marker for native liver survival. Specifically, the PEDFIC2 study523 demonstrated a strong correlation between reductions in serum BA after 6 months of odevixibat treatment and native liver survival for up to 3 years. TFS was 100% among serum bile acid responders, whereas non-responders had higher rates of LT. Notably, all patients who demonstrated a clinical response to pruritus at six months also remained transplant-free. Response stratification revealed that 50% of the patients were non-responders, 36% were responders, and 14% were partial responders. Non-responders were those with <30% reduction of serum BA from baseline, partial responders were those with ≥30% to <70% reduction, and responders who presented a ≥70% reduction from baseline or levels ≤70μmol/L.
Similarly, the INDIGO study,524 which evaluated maralixibat, documented that a BA response, defined as serum BA<102μmol/l or a reduction>75% from baseline, was observed within the first 4 weeks of treatment and was sustained over the long term. Notably, all patients who achieved treatment response survived with their native livers after 5 years of treatment. This response was also associated with improvements in growth and quality of life. These results were confirmed in the MARCH-PFIC phase 3 trial, where maralixibat was shown to significantly reduce pruritus and serum BA in children with PFIC.5 In the BSEP cohort, the least-squares mean reduction in morning ItchRO(Obs) score was −1.7 with maralixibat versus −0.6 with placebo and total serum BA decreased by a mean of 176μmol/L with maralixibat, compared to an increase of 11μmol/L with placebo.
However, more comprehensive data are needed to refine patient selection criteria for IBAT inhibitor therapy. For instance, the INDIGO study524 showed that patients with BSEP deficiency due to biallelic PTM did not respond to maralixibat, and similar patients were excluded from the PEDFIC1522 and PEDFIC2523 studies evaluating odevixibat. Moreover, further research is necessary to clarify the long-term impact of IBAT inhibitors on liver disease progression and native liver preservation.
What genetic alterations are present in PFIC type 3 (MDR3)? What is the function of the mutated gene? Is there a genotype–phenotype correlation in PFIC due to MDR3 deficiency?Recommendation:
- •
Genetic analysis must be performed on the parents and siblings after the diagnosis of the index case. (LoE2, strong recommendation).
The ATP binding cassette subfamily B member 4 (ABCB4) gene, located on chromosome 7q21 encodes the multidrug resistance protein 3 (MDR3).526 MDR3 is expressed in the canalicular membrane of hepatocytes,527 where it functions as a phospholipid translocator.528,529 This translocator plays a critical role in secreting phospholipids into the bile, which neutralizes the detergent effect of BA, thereby mitigating their potential hepatotoxicity.530,531 A defect in MDR3 protein leads to a decreased level of phospholipids within the bile, which in turn exacerbates hepatotoxicity, promotes the crystallization of cholesterol, and ultimately increases the lithogenicity of bile and the development of cholesterol gallstones.532
The spectrum of clinical manifestations resulting from ABCB4 mutations is notably broad.533 The severe end of the phenotypic spectrum is represented by PFIC type 3 (PFIC3), typically associated with biallelic mutations.521,534–536 However, cases of severe cholestatic liver disease in children with monoallelic mutations have also been documented.533,536–538 Defects in MDR3 may manifest as chronic liver disease or cryptogenic cirrhosis in adulthood. Additionally, a range of less severe diseases, commonly linked to monoallelic mutations,539 includes transient neonatal cholestasis,538 low-phospholipid associated cholelithiasis (LPAC),540,541 intrahepatic ICP,542–544 DILI,545 and contraceptive-induced cholestasis. Furthermore, variations in ABCB4 have been implicated in an increased risk of developing hepatobiliary cancer.546
In PFIC3, genotype–phenotype correlations show that more deleterious mutations, such as splice-site, frameshift, and nonsense mutations, which result in truncated proteins or complete absence of residual functional proteins, are frequently associated with a more severe and earlier disease presentation.506,534–536,547 Clinically, these severe mutations are associated with a lack of canalicular MDR3 expression in immunostaining and an absence of response to UDCA, often leading to LT within the first decade of life. However, certain missense mutations that allow residual protein activity533 may result in milder disease phenotypes with a potential response to UDCA.547
What is the clinical spectrum of PFIC3 associated with MDR3 deficiency? How is a diagnosis made? What is the age of onset? What are the typical signs and symptoms of this variant? Is extrahepatic involvement present in these patients? What are the analytical characteristics of these patients?Recommendations:
- •
The diagnosis of severe MDR3 deficiency should be considered in pediatric patients with neonatal cholestasis accompanied by elevated GGT and high serum BA, and in patients with chronic cholestasis appearing during the first year of life, especially if pruritus, hepatomegaly, and splenomegaly are present. (LoE4, strong recommendation).
- •
Surveillance of HCC is recommended. (LoE4, strong recommendation).
- •
A family history of consanguinity, intrahepatic cholestasis of pregnancy in the mother, or confirmed family cases of PFIC3, IPC, or LPAC should increase suspicion of PFIC3. (LoE4, strong recommendation).
- •
The diagnosis of PFIC3 must be confirmed by genetic studies of ABCB4. The presence of biallelic mutations confirms the diagnosis; however, severe disease has also been reported with monoallelic mutations. (LoE3, strong recommendation).
The diagnosis of severe MDR3 deficiency warrants consideration of various clinical presentations.506,533–536,539 The spectrum of manifestations includes neonatal cholestasis, characterized by elevated GGT and serum BA, which may or may not be accompanied by pale stools. This presentation is comparatively rarer than PFIC1 or PFIC2, necessitating a thorough differential diagnosis that includes biliary atresia, Alagille syndrome, alpha 1-antitrypsin deficiency, neonatal sclerosing cholangitis associated with Claudin-1 or DCDC2 deficiencies, and ciliopathies.539
Furthermore, chronic cholestatic liver disease manifesting within the first year of life,533,535,536,539 presenting with symptoms such as pruritus, hepatomegaly, splenomegaly, and jaundice, although jaundice is not invariably present and sometimes pale stools, is also indicative of severe MDR3 deficiency. The intensity of pruritus is usually less pronounced than that of PFIC1 and PFIC2, and laboratory findings often reveal elevated bile acid and transaminase levels.535,536 It is important to exclude other etiologies of cholestasis, and clinicians should be aware that late-onset cases are possible. PH533,535,536,539 and its consequences, such as ascites and gastrointestinal bleeding, can occur in the absence of overt cholestasis in pediatric, adolescent, and young adult patients. This scenario leads to the need to exclude other hepatic conditions that can complicate with PH. In addition, regular surveillance for HCC remains a recommended practice. Notably, no extrahepatic manifestations have been linked to MDR3 deficiency, aligning with its exclusive expression in the canalicular membrane of hepatocytes.535,548,549
Confirmation of PFIC3 diagnosis requires genetic evaluation of ABCB4. Although the presence of biallelic mutations is diagnostic, severe disease phenotypes have also been reported in patients with monoallelic mutations.536,547 Histopathological examination typically reveals ductular proliferation,533,535,536,550 while immunostaining for MDR3 in hepatic tissue can assist in the diagnosis.533,535,536 The absence of canalicular MDR3 expression supports this diagnosis; however, normal staining patterns may still be observed in patients with milder genotypes.533,535
Obtaining complete medical and family history is essential. Information regarding consanguinity533,535,536,566,568,569 confirmed familial cases of PFIC3,535 ICP,533,535,536,539 or LPAC533,535,536,539 can suggest the diagnosis of PFIC3. It is also important to acknowledge that MDR3 deficiency may be less overtly than classical PFIC in children, manifesting as chronic asymptomatic liver dysfunction, transient neonatal cholestasis, or LPAC.
What is the typical clinical course of PFIC3 in children, and how should disease progression be monitored?Recommendations:
- •
Patients with disease progression must be evaluated as candidates for LT. (LoE4, strong recommendation).
- •
The assessment of pruritus at each medical visit is recommended, as well as the impact on sleep, daily activities, and quality of life of the patient and family. (LoE4, strong recommendation).
- •
Cholelithiasis must be investigated during follow-up. (LoE4, strong recommendation).
- •
HCC screening should be performed even in young patients. (LoE4, strong recommendation).
Referral to a tertiary pediatric liver center, where specialized diagnostic and therapeutic care is available, is a key component in the management of pediatric patients with PFIC3. The disease trajectory in PFIC3 patients is often dictated by the functional integrity of MDR3, as those with complete loss of function typically progress to cirrhosis in the first decade of life, accompanied by the onset of PH and liver failure. Thus, regular evaluation of these patients for LT is warranted, particularly in those who exhibit disease progression during the follow up.
The assessment of pruritus539 is a critical component of clinical follow-up and should be thoroughly evaluated at each visit. This includes assessing its impact on sleep, daily activities, and overall quality of life for both patients and family members. Additionally, cholelithiasis should be investigated as part of routine follow-up in patients with PFIC3. Given the increased risk of HCC in PFIC3, even in young patients, systematic screening for HCC is recommended.535,548 The identification of HCC in PFIC3 patients is an immediate indication for LT, reinforcing the importance of this surveillance.
How does UDCA affect the progression of PFIC3?Recommendations:
- •
Treatment with UDCA is recommended for all PFIC3 patients, although the response may vary across different genotypes. (LoE4, strong recommendation).
UDCA therapy533,535,539,548 is recommended for all patients diagnosed with this condition. The rationale for UDCA administration lies in is its ability to modify bile composition, leading to a reduction in its toxicity and lithogenicity. Additionally, UDCA may exert chaperone-like effects, thus facilitating stabilization and functional optimization of the affected proteins involved in bile acid transport. Nevertheless, the dose should not exceed 30mg/kg/day to reduce the risk of adverse effects.
Improvement in native liver survival has been documented in patients who respond positively to UDCA.535 Notably, the genotype of an individual with PFIC3 plays a relevant role in determining the efficacy of UDCA.533,535,536,547 The presence of severe mutations, such as those leading to truncated proteins or complete absence of residual functional proteins, is associated with a lack of response to UDCA. Patients with such genotypic profiles typically require LT within the first decade of life. Conversely, patients with missense mutations that preserve some degree of protein activity may experience a less aggressive disease course. These patients are more likely to respond favorably to UDCA therapy, potentially improving native liver survival outcomes.
What therapeutic alternatives are available for patients with PFIC3?Recommendations:
- •
Nutritional support must be initiated at diagnosis, with formulas enriched in MCT in infants with cholestasis. (LoE4, strong recommendation).
- •
Fat-soluble vitamins must be regularly monitored, and supplementary treatment must be initiated after diagnosis. (LoE4, strong recommendation).
- •
Early treatment with UDCA is recommended. The dose should not exceed 30mg/kg/day. (LoE4, strong recommendation).
- •
IBAT inhibitors may be an option; however, information on PFIC3 is scarce. (LoE4, weak recommendation).
- •
LT must be indicated in cases of end-stage liver disease with PH, liver dysfunction, refractory pruritus, or HCC. (LoE4, strong recommendation).
The initiation of nutritional support is imperative at the time of diagnosis. For infants presenting with cholestasis, formulas with MCT are recommended because absorption is more readily observed in the absence of bile. Moreover, vigilant monitoring of deficiencies in fat-soluble vitamins is crucial,512,513 with the initiation of supplementary treatment if needed.512,513
Additional treatments for pruritus, such as rifampicin and phenobarbital, have been used with varying degrees of efficacy. IBAT inhibitors, recently approved for patients with PFIC, may also have therapeutic potential in pruritus management in patients with MDR3 deficiency. Although, most of the available data on IBAT inhibitors comes from patients with BSEP or FIC1 deficiencies,522–524 an increasing body of evidence is emerging for PFIC3. In this context, a real-world study including patients with various PFIC subtypes, including PFIC3, demonstrated that odevixibat is a safe and effective treatment for reducing serum bile acid levels and improving pruritus.551 Moreover, the MARCH-PFIC trial, which demonstrated the efficacy of maralixibat in managing pruritus in PFIC, included a cohort where nine patients with biallelic MDR3 deficiencies were included, among others.5
In cases where patients progress to end-stage liver disease presenting with PH, liver dysfunction, refractory pruritus, or HCC, LT is indicated.533,539
What is the survival benefit of LT in patients with PFIC?Recommendation:
- •
The relapse of BSEP deficiency after LT should be monitored in every patient during follow-up. (LoE4, strong recommendation).
- •
Risk-benefit evaluation should be balanced prior to LT in patients with FIC1 deficiency, as graft steatosis is almost universal and diarrhea and other extrahepatic symptoms may severely worsen. (LoE4, strong recommendation).
LT is an effective therapeutic intervention for patients with BSEP and MDR3 deficiencies,487,515,552–554 leading to prolonged survival, improved quality of life and catch-up growth. Despite these benefits, clinicians must remain vigilant during the post-transplantation follow-up of patients with BSEP deficiency, as there exists a potential for disease recurrence mediated by alloimmune mechanisms. This risk factor requires careful monitoring.
When considering LT for FIC1 deficiency,552,555 the decision should be meticulously weighed. Although transplantation can address primary liver disease, postoperative challenges are notable, with almost all patients developing graft steatosis.554–557 Moreover, certain extrahepatic symptoms of the disease,556 such as diarrhea555,558,559 and pancreatitis, may exacerbate post-transplant, and patients may experience no improvement in growth and a delay in puberty onset.
Addressing post-transplant steatosis remains an area that requires further research to identify the optimal prevention strategies. Preliminary evidence suggests that SBD560–562 either concurrent with or following LT, is associated with favorable outcomes. In addition, the advent of IBAT inhibitors has presented a novel therapeutic approach. However, the efficacy of IBAT inhibitors in preventing post-transplant steatosis requires further investigation to fully determine their potential benefits and refine therapeutic protocols.
What are the indications for LT in patients with PFIC?Recommendations:
- •
In patients with PFIC1, LT should be indicated in children with cirrhosis or severely impaired quality of life due to pruritus, without response to medical or surgical management. (LoE4, strong recommendation).
- •
In patients with BSEP deficiency, LT must be indicated for those who develop end-stage liver disease or HCC. (LoE4, strong recommendation).
- •
LT should be indicated for children with MDR3 deficiency who develop end-stage liver disease with PH or hepatic insufficiency, refractory pruritus, or HCC. (LoE4, strong recommendation).
In the context of BSEP deficiency,553,563 LT is indicated when patients progress to end-stage liver disease with hepatic insufficiency or develop hepatocarcinoma. Similarly, in PFIC1,563 which often presents during childhood, the development of cirrhosis or severely impaired quality of life due to intractable pruritus that is unresponsive to medical or surgical biliary diversion is an indication for LT. However, it is important to note that post-transplantation outcomes in PFIC1 may be compromised by graft steatosis and persistent or aggravated extrahepatic symptoms, such as diarrhea.
LT should also be considered for patients with MDR3 deficiency,553,563 particularly in children who progress to end-stage liver disease with PH or hepatic insufficiency, refractory pruritus, or HCC.
What is the management of post-transplant diarrhea in patients with PFIC1?Recommendation:
- •
SBD and IBAT inhibitors may be used to treat diarrhea after LT. (LoE5, weak recommendation).
LT in patients with PFIC1 often leads to complex postoperative complications. One of the most notable complications is the exacerbation of diarrhea,555,559 which is likely attributed to the restoration of normal FIC1 function in the liver, whereas the deficiency persists in the intestinal tract, creating a discordance in bile salt processing between these organs. To mitigate post-transplant diarrhea, SBD562 and IBAT inhibitors564 have shown promising results. However, conclusive evidence supporting its efficacy is still lacking, necessitating further research.
What is the management of disease recurrence after LT in patients with PFIC2?Recommendations:
- •
Recurrence of BSEP deficiency after LT should be considered in patients with pruritus, jaundice, increased aminotransferase and bilirubin levels, and normal GGT levels. (LoE4, strong recommendation).
- •
Determination of antibodies against BSEP and liver biopsy are recommended for diagnosis. (LoE4, strong recommendation).
- •
Corticosteroid boluses, anti-CD20, plasmapheresis or immunoadsorption should be considered as potential treatment alternatives. (LoE5 strong recommendation).
PFIC2 recurrence is believed to be immunologically mediated and is characterized by the formation of BSEP-inhibitory antibodies directed against the donor's BSEP protein.511,565,566 Suspicion should be raised when clinical manifestations such as pruritus with or without jaundice and elevated aminotransferase and bilirubin levels with normal GGT levels are observed. The presence of serum antibodies against BSEP and liver biopsy findings, particularly intrahepatic cholestasis and giant cell transformation, is key to confirming the diagnosis.
Management of BSEP deficiency recurrence after LT primarily involves increasing immunosuppression. Treatment modalities, including corticosteroid boluses,565 anti-CD20 therapy,567 plasmapheresis568 or immunoadsorption,569 have been employed successfully in severe episodes, leading to resolution of symptoms.
What is the role of the new variants in genetic cholestasis (TJP2, Farnesoid X Receptor Deficiency, Myosin 5 B Deficiency, Kif 12, ARC syndrome)?Recommendations:
- •
Genetic analyses must be performed to confirm the diagnosis of all autosomal recessive diseases. (LoE4, strong recommendation).
- •
Immunostaining of liver tissue may be helpful in differentiating FXR, BSEP, and TJP2 deficiency. (LoE4, weak recommendation).
- •
Regular surveillance for HCC is indicated at an early age in patients with TJP2 deficiency. (LoE4, strong recommendation).
- •
Nutritional support with fat-soluble vitamin supplementation is indicated in patients with FXR, TJP2, MYO5B, ARC syndrome or KIF 12 deficiency. (LoE4, strong recommendation).
- •
Pruritus can be treated with UDCA, phenobarbital, rifampicin, cholestyramine, and IBAT inhibitors. However, in other types of PFIC beyond types 1, 2, and 3, the role of IBAT inhibitors should be studied further. (LoE4, weak recommendation).
- •
LT is indicated in the presence of advanced liver disease and in cases of hepatocarcinoma with TJP2 deficiency. (LoE4, strong recommendation).
Recent advances in the understanding of the molecular patterns of bile formation have facilitated the diagnosis of previously unclassified cases of neonatal cholestasis,14,22,505 with a predominance in the normal GGT cholestasis category, although such cases remain relatively rare. Deficiencies in Tight junction protein 2 (TJP2), USP53,570 and UNC45571 have been linked to the structural polarity of hepatocytes, whereas Myosin5B (MYO5B) and VPS 33B/VIPAR defects, seen in arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome, disrupt functional polarity. Moreover, defects in the FXR impair regulatory mechanisms governing bile acid synthesis, secretion, and uptake.
Genetic confirmation572 is pivotal for these autosomal recessive diseases, necessitating the identification of biallelic mutations in the following genes: TJP2 for TJP2 deficiency, NR1H4 for FXR deficiency, MYO5B for MYO5B deficiency, along with VPS33B and VIPAR for ARC syndrome, and KIF 12 for KIF12 deficiency. Differential diagnosis is essential to delineate TJP2, FXR, MYO5B, USP53, UNC45, BSEP, and FIC1 deficiencies, with ARC syndrome being distinguishable by its hallmark manifestations along with normal GGT neonatal cholestasis.
Clinically, TJP2,573,574 FXR,575 and BSEP deficiencies should be suspected in infants presenting with normal GGT cholestasis, intractable pruritus, elevated serum BA and aminotransferases, and rapid progression to end-stage liver disease. Notably, extrahepatic manifestations, predominantly respiratory and neurological, are exclusive to TJP2 deficiency. From a pathological standpoint, these conditions also exhibit giant cell transformation in liver biopsies. Immunohistochemical staining aids in differential diagnosis; absent BSEP canalicular expression in FXR and BSEP deficiencies, while in TJP2 deficiency, there is abnormal Claudin-1 localization and absent TJP2 expression. Surveillance for HCC from an early age is indicated for TJP2,576 similar to the approach in BSEP deficiency.
Differentiating MYO5B577–579 from FIC1 deficiencies is crucial, as they both entail normal GGT cholestasis with pruritus, elevated serum BA, and mild aminotransferase increases, coupled with diarrhea and hearing loss as common extrahepatic manifestations. ARC syndrome580–582 should be suspected in infants with normal GGT cholestasis, pruritus, Fanconi syndrome, and arthrogryposis, with accompanying ichthyosis, platelet dysfunction, failure to thrive, recurrent sepsis, and deafness. Liver biopsy may reveal giant cell transformation, and immunostaining may reveal mislocalized BSEP. The prognosis is generally poor, with frequent mortality occurring within the first year.
Differential diagnosis between KIF 12 deficiency583,584 and other high GGT neonatal cholestasis such as biliary atresia or MDR3 deficiency is vital as they share clinical and histological features, including ductular proliferation. KIF 12 deficiency may progress to PH and sclerosing cholangitis.
Treatment for FXR, TJP2, MYO5B, ARC syndrome and KIF 12 deficiencies includes nutritional support and fat-soluble vitamin supplementation. The management of pruritus may include the use of UDCA, phenobarbital, rifampicin, cholestyramine, and antihistamines. The role of IBAT inhibitors is yet to be fully defined, with only limited experience suggesting a potential relief from pruritus. LT is reserved for advanced liver disease or HCC in patients with TJP2 deficiency. Conversely, the multisystemic severity of ARC generally precludes transplantation.
How should we diagnose a FIC1 deficiency in adults? What is the clinical presentation of FIC1 deficiency? How do we monitor the progression of these patients? Is there a genotype–phenotype correlation in FIC1 deficiency? Is it necessary to perform a family segregation study?Recommendation:
- •
In adult patients with persistent cholestasis of unknown origin, it is recommended to perform genetic study of variants associated with cholestatic disease (LoE 5, Strong recommendation).
- •
In adult patients diagnosed with FIC1 deficiency, it is suggested to conduct clinical and analytical follow-up every three to six months, evaluating liver function, nutritional status and symptoms associated with the disease. (LoE5, weak recommendation).
- •
Assessing liver fibrosis every 1–2 years is suggested in these patients. (LoE5, weak recommendation).
- •
Family segregation study should be considered when a patient with adult-onset chronic cholestasis displays a specific pathogenic genetic variant (LoE 5, strong recommendation).
Compared to children, the condition in adults is usually milder. Genetic testing is recommended when no clear cause of cholestasis is identified, and more common forms of adult cholestasis have been ruled out. This analysis should be conducted considering the resources available at the center, as described above. Although FIC1 deficiency can manifest in adulthood, its occurrence at this stage is relatively uncommon. The frequency of cholestasis episodes with jaundice and pruritus, in the setting of BRIC, can be highly variable and may be influenced by external triggers. The term “benign” refers to the absence of liver damage and the lack of progression of fibrosis, but in some cases, it may cause chronic, progressive damage only visible over time. Thus, the use of the term benign should likely be reserved for patients without signs of advanced disease or without progression over time.585–587 As for other liver diseases, regular monitoring of liver fibrosis at intervals of one to two years is suggested.
Limited data is available regarding ATP8B1 variants in heterozygosity in the adult population. A large cohort study of adult patients with genetically associated liver disease found that the residual function of the FIC1 protein determines the clinical phenotype in individuals with biallelic variants in ATP8B1. Most early onset FIC1-deficiency patients had a complete loss of function and those with BRIC had 5–20% of the functional capacity of a healthy subject. Most heterozygous patients exhibit substantially higher levels of FIC1, maintaining at least 50% of functional capacity. Therefore, it has been hypothesized that monoallelic variants of ATP8B1 may significantly contribute to the development of chronic liver disease, rather than being primarily responsible for a classic PFIC1.588
On the other hand, another study showed that some individuals with a single heterozygous variant developed symptoms even before their first birthday, suggesting the involvement of additional mutations in other genes, epigenetic alterations or environmental factors contributing to the severity of the cholestatic phenotype. Notably, the authors found variable patient outcomes associated with the same missense mutation of FIC1. Determining the causal role of many missense variants remains challenging as substantial number of them are classified as VUS. In another study, patients with pathogenic or likely pathogenic (P/PP) mutations exhibited higher liver stiffness and elevated bile acid levels compared to those without causal variants, and all presented histological evidence of lobular cholestasis, confirming a more aggressive phenotype in individuals with causal mutations. Further studies are needed to better understand the long-term clinical implications of mutations in genes associated with intrahepatic cholestasis.494,589–591
In patients with adult-onset cholestasis, the absence of family history of a similar phenotype should not prevent physicians from investigating a genetic cause of an unexplained liver disease. Such conditions may arise from a de novo variant, meaning it is not inherited from either parent, or follow a recessive inheritance pattern, with both parents typically being healthy carriers and 75% of siblings clinically unaffected. The substantial number of causative mutations identified in adults with cryptogenic cholestasis supports the utility of mutational screening particularly in individuals with a history of pruritus, regardless of the age at onset of cholestasis.496,592–594
How should we diagnose a BSEP deficiency? What is the clinical presentation of BSEP deficiency in adults? How do we monitor the progression of these patients? Is there a genotype–phenotype correlation in BSEP deficiency? Is it necessary to perform a family segregation study?- •
In adult patients with persistent cholestasis, it is recommended to perform genetic study of variants associated with cholestatic disease (LoE 5, Strong recommendation).
- •
Family segregation study should be considered when a patient with adult-onset chronic cholestasis displays a specific pathogenic genetic variant (LoE 5, strong recommendation).
As previously described, the diagnosis and evaluation of the disease should be based on genetic testing. The genetic characteristics of this condition are discussed in detail in the pediatric section. From a clinical standpoint, adult patients with BSEP deficiency usually have a different clinical pattern than the classic infantile PFIC2. Chronic, progressive cholestatic liver disease from infancy is uncommon in these adults. Instead, they more frequently have an intermittent or inducible cholestasis phenotype.588 A recent adult cohort study showed that patients with ABCB11 variants typically presented with episodic acute cholestasis and had no evidence of ongoing chronic liver disease between flares. Many such patients were heterozygous for missense ABCB11 mutations, meaning they have partial BSEP function preserved. It appears that if BSEP function is above a critical threshold (20–25% of normal), the liver can remain stable under baseline conditions, but stressors can precipitate cholestatic episodes.588
Although hepatic dysfunction during pregnancy is often associated with ABCB4 mutations, one study identified that 82% of adult women with ABCB11 variants presented with pregnancy-associated liver dysfunction as the initial manifestation of the disease.588 Affected women develop pruritus and cholestatic labs during pregnancy, which resolve after delivery. Outside of pregnancy, both men and women with partial BSEP defects can experience BRIC2. In one series, about 40% of adults with ABCB11 variants had a history of acute or episodic cholestasis unrelated to pregnancy.588 These flares may be set off by other environmental factors such as certain medications (especially those known to inhibit bile transport), infections, or other stressors. Symptoms during cholestatic episodes in adults mirror those seen in pediatric PFIC, though often less severe.
What are the clinical characteristics of adult patients with ABCB4 variants?Recommendation:
- •
Presence of hepatolithiasis before age of 40, hyperechoic intrahepatic foci and recurrence of symptoms after cholecystectomy should rise the suspicion of LPAC. (LoE 4, strong recommendation).
- •
ABCB4 gene sequencing is not required for confirming the diagnosis of LPAC. However, it may help to classify patients with LPAC related to ABCB4 mutations. (LoE5, strong recommendation).
Variants in ABCB4 gene results in a diversity of phenotypic manifestations during adulthood, including chronic cholestasis, ICP, DILI, LPAC syndrome and HCC.595 Depending on the clinical presentation, the disease may follow an asymptomatic or minimally symptomatic course, or it may adopt a more aggressive pattern, characterized by early development of cirrhosis and PH. Histologically, the presence of biliary fibrosis with ductal proliferation, macrophage infiltration in the portal spaces with the presence of giant cells, ductopenia and cholesterol crystals has been documented.596,597
LPAC presentation is more prevalent in females and is characterized by the premature occurrence of intrahepatic cholelithiasis, generally before the age of 40.540 In addition, it has been identified that approximately 15% of cases of ICP are associated with mutations in ABCB4.598
What diagnostic strategies are appropriate for identifying the diverse phenotypes linked to ABCB4 mutations?- •
In adult patients with persistent cholestasis, it is recommended to perform genetic study of variants associated with cholestatic disease (LoE 5, Strong recommendation).
- •
Family segregation study should be considered when a patient with adult-onset chronic cholestasis displays a specific pathogenic genetic variant (LoE 5, strong recommendation).
In patients with chronic cholestasis of indeterminate etiology, it is imperative to exclude the presence of other diseases associated with mutations in ABCB4, particularly when complementary studies yield inconclusive results. These patients typically exhibit a marked elevation in GGT levels; however, normal GGT values do not preclude the diagnosis.599 Histopathological examination frequently reveals cholesterol crystals within the hepatic parenchyma indicative of impaired function of the phospholipid transporter MDR3.600
Concerning LPAC, suspicion should arise in patients presenting with hepatolithiasis who fulfill two of the following criteria: (a) symptom onset before the age of 40; (b) presence of hyperechoic intrahepatic foci, biliary sludge, or intrahepatic microlithiasis; (c) persistence or recurrence of symptoms following cholecystectomy.601 In an appropriate clinical context, alongside the characteristic symptomatology of biliary colic, the identification of twinkling echographic artifacts associated with lithiasis, corresponding to the ultrasound comet-tail sign, is common. Although not essential for diagnosis, genetic testing is necessary to confirm LPAC in the context of ABCB4 mutations, as there are no definitive clinical, analytical, or radiological features that are exclusively associated with mutations in this gene.
What is the typical clinical course of ABCB4 variants in adults? What strategies are recommended for monitoring affected patients?Recommendation:
- •
Hepatobiliary neoplasia screening could be considered in patients with mild symptomatic MDR3 variants diagnosed during adulthood. (LoE 5, weak recommendation).
- •
In patients diagnosed with LPAC without advanced fibrosis or cirrhosis, liver cancer screening could be considered. (LoE5, weak recommendation).
Contrary to patients diagnosed with PFIC3 in childhood, adults usually present a less symptomatic clinical course. Adults tend to experience less intense pruritus, which is a virtually universal symptom in pediatric patients. Additionally, there is an association with a variable prevalence of jaundice, a heterogeneous risk of lithiasic biliary complications and a lower prevalence of PH at the time of diagnosis, resulting in a lower need for LT.602–604 However, according to some studies, individuals heterozygous for ABCB4 may have an increased risk of developing fibrosis/cirrhosis, but further studies are necessary. Additionally, there is a variable risk of developing liver cancer. Although an increased risk of developing CCA had been described,605 a recent work reported a limited risk of CCA in these patients.606 Nevertheless, it is worth mentioning that a higher incidence in hepatobiliary neoplasms has been described in ABCB4-related LPAC compared to non-ABCB4 LPAC.607 Accordingly, regular screening for liver cancer should be considered.
On the other hand, the prognosis for patients diagnosed with LPAC is usually benign, without a defined higher risk of serious complications or neoplastic transformation. These mutations can cause adult-onset recurrent benign intrahepatic cholestasis with slow liver disease progression or symptoms, or biochemical changes related to cholestasis. Furthermore, it is speculated that gene variants may exacerbate liver disease caused by other factors or increase the risk of liver disease progression, but further research is necessary to confirm these hypotheses.
What is the impact of UDCA in the evolution and prognosis of this variant in adults?Recommendations:
- •
The use of UDCA is recommended in patients with ABCB4 mutations and those diagnosed with LPAC (regardless of the presence of ABCB4 mutations). (LoE 4, strong recommendation).
UDCA is the cornerstone in the treatment of the clinical variants linked to mutations in the ABCB4 gene. However, the evidence of its efficacy in adults is limited.608,609 It is important to emphasize that the type of mutation in ABCB4 influences the therapeutic response, with a better response noted in patients with missense mutations compared to those with premature stop codon mutations.
What therapeutic alternatives are available for these patients?Recommendations:
- •
Cholestyramine, fibrates and rifampin may be indicated for pruritus management. (LoE5, weak recommendation).
For patients with refractory pruritus or those who only show a partial response, rifampicin, fibrates and cholestyramine may be considered as potentially effective alternatives. However, the available evidence is still insufficient to establish a specific recommendation. From a pathophysiological standpoint, PPAR agonists, such as bezafibrate and fenofibrate, could be effective, although the limited information available prevents any recommendation in this regard.
Regarding the management of LPAC syndrome, obeticholic acid has been reported to be a viable therapeutic option for those patients who do not tolerate or respond to UDCA. Nevertheless, data on this alternative are limited to a small number of clinical cases, which restricts the ability to draw generalizable conclusions.610
At what age does BRIC1/2 typically begin? What are the common signs and symptoms associated with this variant? Do these patients experience any extrahepatic involvement? What are the analytical features observed in these individuals? How does this variant progress clinically, and what methods do we use to track the progression in these patients?Recommendations:
- •
In patients without symptoms, clinical and laboratory follow up may be done once a year. (LoE 5, weak recommendation).
- •
In patients with symptoms and during acute episodes, the clinical, radiology and laboratory follow up must be adapted to the clinical situation of each patient. (LoE 5, weak recommendation).
BRIC 1 is an inherited disease with autosomal recessive transmission and variable penetrance. It originates as a result of pathogenic mutations in the ATP8B1 gene.490,492 This genetic alteration causes an imbalance between the absorption and secretion of BA,611 as a consequence of which patients experience self-limiting episodes of pruritus and cholestasis. This disease is usually diagnosed in adolescence or around the second decade of life and, although scientific evidence is limited, it appears not to be associated with extrahepatic disease.612 The period between episodes is marked by both clinical and analytical normalcy, which may persist for several months or even years. During episodes, the most common symptoms are jaundice and pruritus and patients may also present with steatorrhea, weight loss and bleeding (as a consequence of malabsorption of fat-soluble vitamins). From a biochemical point of view, these patients show significant elevations in alkaline phosphatase and bilirubin, with normal or slightly elevated transaminase levels and normal or even low GGT.
This disease does not present with extrahepatic involvement, although a higher incidence of cholelithiasis has been described. Analytically, they present a profile very similar to BRIC1, with marked elevation of alkaline phosphatase, normal GGT and variable transaminase levels.
Is it necessary to treat patients with BRIC1/2?Recommendations:
- •
UDCA should be considered to manage symptoms during acute episodes. (LoE5 strong recommendation).
- •
Maintaining treatment between episodes is controversial and should be discussed with the patient. (LoE5, weak recommendation).
The initial treatment for managing the episodes of symptomatic disease is usually UDCA.613 However, patients often experience only a partial response to the drug. Therefore, other therapeutic alternatives have been explored that have demonstrated variable efficacy, including cholestyramine, rifampicin and phenobarbital.613 Nonetheless, there is a lack of evidence regarding the appropriate approach to intercrisis therapeutic management or its necessity. The decision on treatment should be evaluated based on individual clinical manifestations, the frequency of episodes and patient preferences.
What is the role of the new variants in genetic cholestasis (TJP2, Farnesoid X Receptor Deficiency, Myosin 5B Deficiency, Kif 12, ARC syndrome)? What is the role of inborn errors in bile acid synthesis (3β-HSD and AKR1D)?Some clinical cases of patients with cholestasis as a consequence of a mutation in TJP2 have been described. Information is scarce and although initial studies reported normal GGT in patients with this mutation,614 other studies have described the same alteration associated with elevated GGT levels.615
Intrahepatic cholestasis of pregnancyWhat clinical and analytical findings raise suspicion of ICP? How is this condition diagnosed?Recommendation:
- •
In pregnant women who present with pruritus during the second or third trimester of pregnancy, conducting a bile acid study to assess the presence of ICP is recommended. (LoE 4, strong recommendation).
ICP is a liver disorder unique to pregnancy. Clinical suspicion typically arises from the onset of pruritus during the second or third trimester.616 The most used diagnostic biomarkers in clinical practice are total serum BA levels, either alone or in conjunction with elevated transaminase levels. In fact, the characteristic biochemical profile of ICP includes an increase of both.617 The definitive diagnosis of ICP relies on the combination of pruritus in a pregnant woman and a serum bile acid concentration exceeding 10μmol/L.618,619
What impact does ICP have on pregnancy?Recommendation:
- •
It is suggested to perform frequent blood pressure monitoring and close follow-up by the healthcare team monitoring the pregnancy due to an increased risk of preeclampsia, preterm birth and fetal death, among others. (LoE 2, strong recommendation).
ICP is associated with an increased risk of gestational diabetes and preeclampsia, particularly in women with moderate (defined by total serum bile acid levels ≥40μmol/L) or severe ICP (levels ≥100μmol/L).620–622
Fetal morbidity and mortalityAlthough data remain controversial, the most recent evidence, derived from an individual participant data systematic review and meta-analysis of patients with ICP, indicates that the risk of stillbirth is significantly higher than that in the general population when total serum bile acid levels are ≥100μmol/L.622,623
While the exact pathophysiological mechanisms underlying fetal death in ICP are not fully understood, it is hypothesized that elevated BA exert an arrhythmogenic effect on the fetal myocardium, potentially contributing to sudden intrauterine death.624,625
A recent meta-analysis including over 5000 women with ICP demonstrated that both moderate (bile acid levels 40–99μmol/L) and severe ICP (≥100μmol/L) are associated with increased risks of preterm birth, both spontaneous and iatrogenic, as well as the presence of meconium-stained amniotic fluid and a greater need for neonatal intensive care unit (NICU) admission.622 Additional studies have linked ICP to an elevated risk of cesarean delivery and neonatal respiratory distress syndrome.626,627
What is the recommended treatment for pregnant women with ICP? What impact does treatment have on maternal and fetal outcomes?Recommendation:
- •
UDCA is recommended as the first-line treatment in patients with ICP. (LoE 2, strong recommendation).
- •
In patients with pruritus who do not respond to UDCA, addition of rifampicin is suggested. (LoE 4, strong recommendation).
The first-line treatment of choice for ICP is UDCA, which has been shown to alleviate maternal pruritus and improve liver biochemistry.628 S-adenosylmethionine has demonstrated modest benefits in reducing pruritus and improving hepatic laboratory parameters, although the supporting evidence is limited to small-scale studies.629 Cholestyramine has also shown efficacy, albeit to a lesser extent than UDCA.630
In pregnant women who exhibit an inadequate response to UDCA, combined treatment with rifampicin has been associated with reductions in both serum bile acid concentrations and pruritus, with a more modest impact on hepatic biochemical markers.616 An ongoing international randomized controlled trial is currently evaluating the efficacy of rifampicin (300mg every 12h) versus UDCA (450–2000mg/day) in women with severe pruritus from the 14th week of gestation and with total serum bile acid levels ≥40μmol/L.631 Glucocorticoids such as dexamethasone have not demonstrated clinical benefit, as the only available randomized trial failed to show significant improvement.632 Experience with fenofibrate remains extremely limited, with partial benefit reported in just three pregnancies involving two women who received the drug from week 30 of gestation onward.185
Beyond its maternal benefits, treatment with UDCA has also been associated with improved fetal outcomes, including reductions in the incidence of preterm delivery, stillbirth, fetal distress, and the need for NICU admission.628,633 These benefits appear to be more pronounced in women with total serum bile acid concentrations ≥40μmol/L.633
What should be monitored during pregnancy in patients with ICP?Recommendations:
- •
In all patients with altered liver biochemistry during pregnancy, the presence of other liver diseases should be ruled out. (LoE 3, strong recommendation).
- •
In patients with suspected ICP, abdominal ultrasonography is recommended to rule out obstructive causes that justify the clinical situation (LoE 3, strong recommendation).
- •
It is recommended to monitor BA levels from week 32, with weekly frequency from that point onwards (LoE 2, strong recommendation).
During the initial diagnosis of ICP, alternative causes of liver disease must be excluded. This includes abdominal ultrasound to rule out the obstructive causes of pruritus. The most reliable prognostic marker for assessing pregnancy-related risk is the total serum BA concentration,622 which should be measured regardless of fasting status.
A serum bile acid level ≥40μmol/L is associated with an increased risk of fetal complications, including prematurity and fetal distress. Values ≥100μmol/L, particularly from the 35th week of gestation onward, have been linked to an elevated risk of stillbirth, warranting the consideration of delivery at that stage.
In cases where ICP is diagnosed prior to the 32nd week of gestation, bile acid levels should be monitored every 1–3 weeks and weekly thereafter. Importantly, the prognostic threshold is defined as the highest value recorded at any point during follow-up; a single bile acid concentration ≥100μmol/L is sufficient to establish a high risk of fetal death and eliminates the need for further serial determinations. Notably, treatment with UDCA does not interfere with the predictive value of this parameter.622,633
Should a “scheduled delivery” be recommended for pregnant women with ICP?Recommendations:
- •
In patients with ICP and BA concentrations between 40μmol/L and 99μmol/L, labor should be induced between weeks 37 and 40. (LoE 2, strong recommendation).
- •
In pregnant women with concentrations higher than 100μmol/L, labor should be induced at week 36. Additionally, if patients present with intractable pruritus or have a history of miscarriage due to this condition, labor should be induced before week 36 (LoE 2, strong recommendation).
In patients with bile acid levels<100μmol/L, the risk of fetal death ranges from 0.13% to 0.4%, which is comparable to that in the general obstetric population. However, when BA levels exceed 100μmol/L, the risk increases significantly, up to 6.8%.634
In clinical practice, labor is often electively induced to prevent adverse fetal outcomes or to manage intolerable maternal pruritus. The rate of induced deliveries varies with the degree of cholestasis: 10.8% in women with BA levels<40μmol/L, 21.6% in those with levels between 40 and 99μmol/L, and 35.8% when BA are ≥100μmol/L.622,634
In general, preterm delivery (<37 weeks) should be avoided in the absence of elevated serum BA.622,634,635 Delivery is typically recommended between 37 and 40 weeks of gestation for patients with BA levels of 40–99μmol/L. In contrast, for women with levels ≥100μmol/L, induction of labor is advised at 36 weeks or earlier if there is severe intractable pruritus, worsening hepatic function, or a history of adverse fetal outcomes in previous pregnancies related to ICP.
What follow-up should be done after childbirth? Can oral contraception be prescribed after delivery?Recommendations:
- •
In patients with a definitive diagnosis of ICP, biochemical normalization should be confirmed 6–8 weeks postpartum (LoE 5, weak recommendation).
- •
Hormonal contraception is not contraindicated in patients with ICP without a history of contraceptive-associated cholestasis (LoE 5; weak recommendation).
ICP is a reversible cholestatic disorder that typically resolves spontaneously after delivery. Pruritus usually subsides at the time of birth or within subsequent days, and hepatic biochemistry (including bile acid concentrations) returns to normal within a few weeks. Biochemical normalization should be confirmed 6–8 weeks postpartum.636
In cases where pruritus persists beyond delivery, a full reassessment of liver disease and bile acid levels is warranted to exclude underlying chronic hepatobiliary disease.636 Combined hormonal contraceptives may be safely used in women with a history of ICP, provided that they have no prior history of cholestasis induced by hormonal contraception. Although theoretical concerns exist, the risk of recurrent cholestasis due to hormonal contraceptive use in these patients is considered low and does not contraindicate their use.636,637
What is the risk of ICP development in future pregnancies?Recommendation:
- •
It is recommended to inform patients with ICP of the risk of recurrence in future pregnancies, as well as the potential risks associated with the disease (LoE 5, strong recommendation).
Women who have experienced ICP are at risk of recurrence in subsequent pregnancies; however, the exact magnitude of this risk remains unknown. It is important to counsel patients that recurrence is possible, even though the precise probability has not been clearly established.638
How should itching be managed during pregnancy?Recommendation:
- •
It is recommended to reinforce the use of non-pharmacological measures as the first treatment option. (LoE 5, strong recommendation).
- •
The use of cholestyramine and/or rifampicin is recommended for the treatment of pruritus in patients who do not respond to nonpharmacological measures. (LoE 2, strong recommendation).
The first-line approach to managing pruritus during pregnancy consists of general non-pharmacological measures, that are inherently risk-free.639 Patients should be advised to avoid exposure to heat, contact with hot or ice-cold water, alcohol consumption, wool clothing, skin friction, and psychological stress. Conversely, the recommended practices include the use of alkaline soaps, short showers or baths with lukewarm water, application of moisturizers or 2% mentholated emollients, wearing loose-fitting cotton garments, and maintaining short fingernails to reduce excoriation.
Among pharmacological options, rifampicin is considered safe for use during pregnancy.180,616 Although evidence regarding cholestyramine is limited, available data suggest that it does not pose a significant maternal–fetal risk.640 In contrast, no safety data are currently available for colestipol or IBATs in pregnant women. Antihistamines such as hydroxyzine and cetirizine are generally regarded as safe.641 Fibrates, despite their teratogenic potential,181 have been used in the late stages of pregnancy for pruritus associated with intrahepatic cholestasis.185
Therefore, although the evidence remains incomplete, rifampicin and cholestyramine may be recommended for the management of pruritus in pregnant patients with cholestatic liver disease. In severe and refractory cases, plasmapheresis has been employed as a rescue intervention.642
Vanishing bile duct syndromeHow is vanishing bile duct syndrome diagnosed?Recommendation:
- •
In cases of intrahepatic cholestasis, a liver biopsy is recommended to establish the diagnosis of vanishing bile duct syndrome (VBDS), once other causes of cholestasis have been excluded. (LoE2, strong recommendation).
VBDS encompasses a heterogeneous group of rare but potentially life-threatening disorders, characterized by persistent intrahepatic cholestasis and progressive loss of interlobular bile ductules.643 This condition can arise in association with a variety of etiologies, including graft-versus-host disease, PBC, PSC, paraneoplastic syndromes (particularly lymphomas), Alagille syndrome, and DILI. In some cases, VBDS occurs without an identifiable underlying cause, and is considered idiopathic.643,644
A definitive diagnosis requires liver biopsy with histological confirmation of bile duct loss in at least 50% of portal tracts in a specimen containing a minimum of 11 portal tracts.643–645 Beyond diagnosis, liver biopsy provides important prognostic information. The presence of elevated serum bilirubin, hepatocellular cholestasis, foam cells, and advanced liver fibrosis (F3–F4) has been associated with increased risk of mortality and the need for LT, with hepatocellular cholestasis identified as the strongest predictor of adverse outcomes.644 These prognostic patterns have been observed both in DILI-associated VBDS and in VBDS of other etiologies.644,645
Timely diagnosis of VBDS is critical, as it enables closer clinical monitoring, informs prognosis, and may guide the use of anticholestatic therapies. Early identification also allows for the implementation of supportive care strategies and improved anticipation of disease-related complications.646
Is UDCA treatment useful in patients with vanishing bile duct syndrome?Recommendations:
- •
Based on current evidence, treatment with UDCA is not recommended in patients with VBDS, regardless of the underlying etiology, as no proven clinical benefit has been demonstrated. (LoE 4, weak recommendation).
Although isolated reports have suggested a potential benefit of UDCA in patients with VBDS647 and its efficacy has been proposed in cases where VBDS is associated with PBC,646 no controlled studies have evaluated its use. Data from the Drug-induced Liver Injury Network (DILIN) registry identified 26 biopsy-confirmed cases of VBDS. Compared with DILI cases without VBDS, these patients exhibited more severe clinical features, including a higher frequency of jaundice (96%), more pronounced cholestasis, increased incidence of fever (46% vs. 25%), higher peak serum bilirubin levels (21.5 vs. 13.9mg/dl), a greater proportion with an R value<2 (57% vs. 23%), and a longer duration to achieve bilirubin levels below 2.5mg/dl. Despite more frequent use of steroids and UDCA in the VBDS group, no therapeutic benefit was observed.644
The severity of bile duct paucity has emerged as a potential prognostic factor, particularly in DILI-associated VBDS, where the mortality rate reached 27%, compared to 9% in patients with DILI not meeting VBDS criteria.645 Across various VBDS etiologies, UDCA has not demonstrated superiority over observation in improving outcomes. Poor prognosis has been consistently linked to elevated serum bilirubin, hepatocellular cholestasis, the presence of foam cells, and advanced liver fibrosis.644
Although UDCA might offer some value in specific cholestatic conditions associated with VBDS, such as PBC, where its benefit has been established,646 current evidence does not support its general use in VBDS.
Extrahepatic complications of cholestatic diseasesHow should we screen and treat bone disease in patients with cholestatic liver disease?Recommendations:
- •
The evaluation of bone mineral density using dual energy X-ray absorptiometry (DEXA) should be performed at diagnosis and every 2–5 years during the follow-up depending on the degree of cholestasis and on an individualized basis. (LoE 4, strong recommendation).
- •
Patients with a femur T-Score<−1.5 should receive treatment following osteoporosis clinical guidelines. (LoE 4, strong recommendation).
- •
Supplements of calcium (1000–1200mg/day) and vitamin D (400–800YIU/day) can be considered but are not recommended in patients with normal nutritional status and lack of features of calcium malabsorption. (LoE 4, strong recommendation).
- •
Bisphosphonates are safe and effective in PBC patients and should be considered to treat patients with a femur T-Score<−1.5 and following osteoporosis clinical guidelines. (LoE 4, strong recommendation).
- •
In patients with esophageal varices consider treatment with parenteral bisphosphonates (pamidronate, ibandronate or zolendronic acid). (LoE 4, strong recommendation).
Patients with long-standing cholestasis have a higher risk of developing osteoporosis. Different studies have shown that osteoporosis is more prevalent in women with PBC than in the general population.648–651 Furthermore, compared with healthy controls, even non-cirrhotic PBC patients have a significantly increased risk of osteoporosis.651 Different factors favoring the loss of bone mass in this liver disease have been described, including advanced age, body mass index, menopausal status, duration of the disease, advanced histological stage and vitamin D deficiency.648–652 Moreover, lobular cholestasis, evaluated as aberrant expression of keratin 7 in hepatocytes, has shown significant negative correlations with bone formation and resorption, indicating low bone turnover and suggesting that this feature of PBC may cause secondary osteoporosis in these patients.651
Apart from the greater prevalence of osteoporosis, the prevalence of sarcopenia, vertebral fractures and osteosarcopenia also seems to be increased and it has been observed that these complications are closely interrelated.650 Fractures, particularly vertebral fractures, are associated with osteoporosis, osteopenia and T scores less than −1.5, while osteoporosis and osteopenia are associated with the severity of liver damage.652
To prevent and treat osteoporosis,43 it is recommended to perform a DEXA at the time of diagnosis and re-evaluate after 1 to 5 years of follow-up, depending on the degree of cholestasis and on an individualized basis. Additionally, a balanced diet, regular physical exercise, and smoking cessation are advised.
Regarding pharmacological treatment, calcium (1000–1200mg/day) and vitamin D (400–800YIU/day) supplements may be considered. Although these supplements are frequently provided, there is no conclusive evidence to support or refute their efficacy. However, they are not recommended for patients with normal nutritional status and no signs of calcium malabsorption. Bisphosphonates, particularly weekly alendronate and monthly ibandronate, have been shown in several trials to effectively increase bone mass in patients with PBC. While these drugs are generally safe and effective, caution is advised in patients with esophageal varices, as they can cause gastritis or esophagitis. In such cases, parenteral bisphosphonates, including pamidronate, ibandronate, or zoledronic acid, may be preferable. Furthermore, hormone replacement therapy has been found to be effective in postmenopausal patients. There is no agreement on the appropriate time to start treatment; however, it seems reasonable to treat patients with femur T-Score<−1.5, following osteoporosis clinical guidelines.43
How is the management of fat-soluble vitamin malabsorption in patients with cholestatic disease?Recommendation:
- •
Fat-soluble vitamins status should be determined in all patients with cholestatic diseases by appropriate blood tests. (LoE4, strong recommendation).
- •
Vitamin supplements should be given in case of vitamin deficiency. In case of vitamin supplementation, it is advisable to periodically monitor vitamin levels to prevent overdose. (LoE4, strong recommendation).
- •
In case of jaundice, routine measurement of vitamin levels is recommended. (LoE 4, strong recommendation).
Patients with chronic cholestasis may present malabsorption of fat-soluble vitamins due to the changes in BA pool. However, deficiencies of fat-soluble vitamins A, D, E and K are rare in patients with chronic cholestasis, with fat-soluble vitamin deficiencies more likely to occur in patients with jaundice, long-lasting cholestasis, or in those patients with osteomalacia.653,654
So, the suggestion is to determine the status of fat-soluble vitamins in all patients with cholestatic diseases by appropriate blood tests to determine the need for supplementation of these vitamins.
Vitamin status should be assessed through specific biochemical measurements, with serum retinol used to determine vitamin A levels, serum 25-hydroxyvitamin D to evaluate vitamin D status, and plasma α-tocopherol to detect vitamin E deficiency. Since no standardized method exists for assessing vitamin K status, it should be determined using a combination of biomarkers alongside dietary intake.655
Specific vitamins, including A, D, E and K should be administered only to patients with confirmed deficiencies. In such cases, periodic monitoring of vitamin levels is advisable to assess treatment effectiveness and prevent overdose.655 For vitamin A deficiency, a dose of 50,000YIU every 15 days is recommended, with regular monitoring to avoid hypervitaminosis A, which can lead to symptoms such as asthenia, lethargy, abdominal discomfort, anorexia, skin peeling, alopecia, intracranial hypertension and hepatotoxicity. Vitamin D supplementation should be at least 1000YIU (25μg) per day, with careful monitoring to ensure efficacy and prevent overdose, which may result in hypercalcemia and hypercalciuria. Unlike the pediatric population, only a minority of adults with cholestasis and low vitamin E levels develop psychomotor and neurological disorders due to vitamin E deficiency.656 However, due to the potential but unrecognized benefits, it is recommended to administer at least 15mg of α-tocopherol per day, particularly in patients with neurological symptoms of uncertain etiology.
The administration of vitamin K in cholestatic diseases remains controversial.657,658 If prolonged prothrombin time is detected due to vitamin K malabsorption, correction can be achieved with 10mg/day of subcutaneous vitamin K for three days, followed by chronic oral supplementation (5–10mg/day) or subcutaneous administration (10mg/month). Additionally, prophylactic vitamin K supplementation should be considered in cases of severe cholestasis before invasive procedures and in the context of bleeding episodes. For some malnourished patients, the administration of MCT in the form of coconut oil extract (one tablespoon three to four times per day) may be beneficial.
Although there are no obvious clinical signs of deficiency, plasma antioxidant levels are low in cholestatic patients even in early stages of the disease. This is probably due to malabsorption of fat-soluble vitamins as well as other hepatic release mechanisms, suggesting the need for dietary supplementation.659
Conflict of interestADG: lecture fees from Ipsen, Gilead, AdvanzPharma, GSK and EISAI; consultant fees from Ipsen, Gilead, GSK and Mirum; travel expenses from Ipsen and AdvanzPharma. CM: none. EGD: lecture fees from Ipsen, Gilead, AdvanzPharma; consultant fees from Ipsen, Gilead, AdvanzPharma. GMB: none. JA: lecture fees from Abbvie, AstraZeneca, Gilead, IPSEN, Madrigal, MSD, Roche; consultant fees from Boehringer, IPSEN, Mirum, MSD, NovoNordisk, Orphalan; grants from Gilead, Orphalan. CFR: lecture fees from Gilead, AdvanzPharma, consultant fees from AdvanzPharma, JQ: consultant fees from Mirum, Ipsen, AdvanzPharma, grants from Ipsen, Mirum. EM: lecture fees from Gilead, Ipsen; consultant fees from AdvanzPharma. MS: lecture fees from Ipsen, Mirum; consultant fees from Ipsen, Mirum, Astellas. AO: consultant fees from AdvanzPharma, Mirum, Ipsen. RM: lecture fees from Gilead, AdvanzPharma, Abbvie. MAD: none. MC: lecture fees from Gilead, Ipsen, Orphalan. LGB: consultant fees from AdvanzPharma. SRT: lecture fees from Gilead, Ipsen, AdvanzPharma; consultant fees from Ipsen, Mirum, grants from Mirum. FJ: lecture fees and travel expenses from Abbvie, Gilead, Casen Recordati. MB: consultant fees from Astellas, Chiesi, Ipsen, Grifols, Orphalan; grant from Gilead. JLM: none. MCL: lecture fees from Gilead, Ipsen, GSK, AdvanzPharma; consultant fees from Gilead, Ipsen, Falk, GSK; grants from Gilead, Mirum, travel expenses from Ipsen, AdvanzPharma.
Uncited referencesVanesa Bernal, Rosa Roca, Juan Turnes, Judith Gómez, Raúl Andrade
Ruth Díez, Ismael El Hajra, Diana Horta, María Mercadal, Inmaculada Castello, Anna Reig, María Rubio, Ana Arencibía, Sara Lorente, Sonia Blanco, Enrique Medina, Rafael González de Caldas, Loreto Hierro, Manuel Hernández, Inés Sáenz de Miera, Mercè Vergara, Carmen Álvarez-Navascúes, Montserrat García-Retortillo, Agustín Castiella
Jose Antonio Martinez Oton, Javier Martínez González, Víctor Vargas, Inés Loverdos
Begoña Polo, María Legarda, Elena Aznal
The followings are the supplementary data to this article:

