







La concentración sérica de fósforo oscila entre 2,5 y 4,5mg/dl (0,81-1,45mmol/l) en adultos, con niveles más altos en la infancia, la adolescencia y durante la gestación. El fosfato intracelular está implicado en el metabolismo intermediario y otras funciones celulares esenciales, mientras que el extracelular es fundamental para la mineralización de la matriz ósea. La fosforemia se mantiene en un estrecho rango mediante la regulación de la absorción intestinal, la redistribución y la reabsorción tubular renal de fósforo. La hipofosfatemia y la hiperfosfatemia son situaciones clínicas frecuentes, aunque, en la mayoría de las ocasiones, se trata de alteraciones leves y poco sintomáticas. Sin embargo, pueden presentarse cuadros agudos y severos que requieren tratamiento específico. En este documento elaborado por miembros del Grupo de Trabajo de Metabolismo Mineral y Óseo de la Sociedad Española de Endocrinología y Nutrición se revisan los trastornos del fosfato y se proporcionan algoritmos de manejo clínico de la hipofosfatemia y la hiperfosfatemia.
Serum phosphorus levels range from 2.5 and 4.5mg/dL (0.81-1.45 mmol/L) in adults, with higher levels in childhood, adolescence, and pregnancy. Intracellular phosphate is involved in intermediary metabolism and other essential cell functions, while extracellular phosphate is essential for bone matrix mineralization. Plasma phosphorus levels are maintained within a narrow range by regulation of intestinal absorption, redistribution, and renal tubular absorption of the mineral. Hypophosphatemia and hyperphosphatemia are common clinical situations, although changes are most often mild and oligosymptomatic. However, acute and severe conditions that require specific treatment may occur. In this document, members of the Mineral and Bone Metabolism Working Group of the Spanish Society of Endocrinology and Nutrition review phosphate disorders and provide algorithms for adequate clinical management of hypophosphatemia and hyperphosphatemia.
El fósforo representa el 1% del peso corporal total. La reserva corporal de fósforo orgánico es básicamente ósea (85%), en forma de cristales de hidroxiapatita; la mayoría del resto de los fosfatos son intracelulares (14%) y menos del 1% del fósforo está en el líquido extracelular. El fosfato circulante existe como especie hidrogenada univalente (4 veces más común) o divalente. La concentración sérica normal de fósforo oscila entre 2,5 y 4,5mg/dl (0,81-1,45mmol/l) en adultos, con niveles más altos en la infancia, la adolescencia y durante la gestación. El fosfato intracelular está implicado en el metabolismo intermediario y otras funciones celulares esenciales, mientras que el extracelular es fundamental para la mineralización de la matriz ósea1,2. Los niveles séricos de fósforo se mantiene en un estrecho rango mediante cambios interrelacionados en la absorción intestinal, la redistribución entre compartimentos (intracelular, extracelular y óseo) y la reabsorción tubular renal1–4.
Los alimentos más habituales de la dieta (lácteos, legumbres, cereales, carnes, pescados, bebidas refrescantes, etc.) son ricos en fósforo (más biodisponibles los de origen animal) y el aporte dietético y fracción absorbida suele ser superior al requerimiento diario (580-1.055mg/día). El 60-70% del fosfato de la dieta se absorbe en el intestino delgado (especialmente en el yeyuno) mediante vías dependientes e independientes del sodio que pueden implicar a la vitamina D1,3.
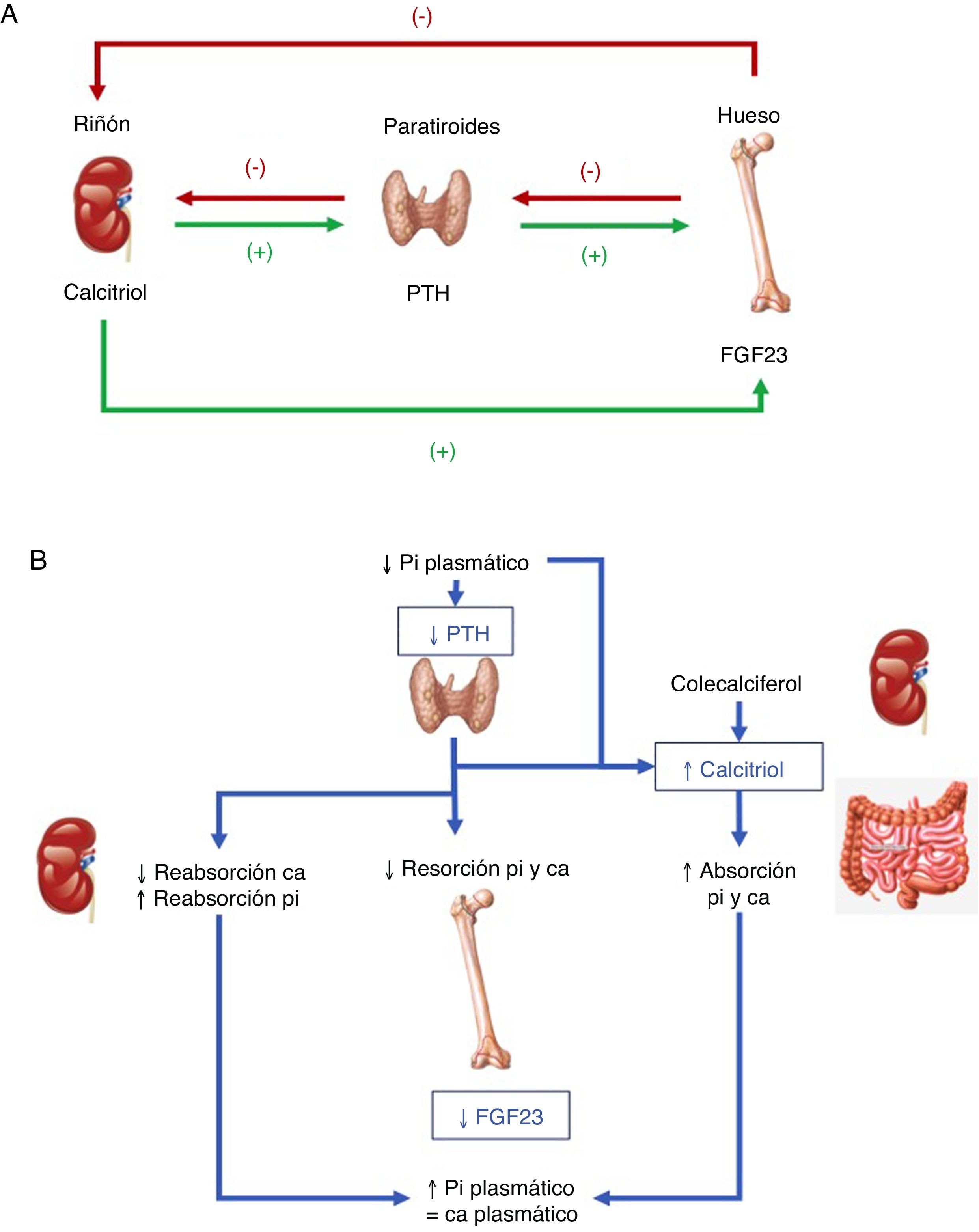
El reservorio óseo de fosfato se moviliza de forma similar al del calcio, aunque es dirigido, además de por la parathormona (PTH) y la vitamina D, por otras hormonas, como el factor de crecimiento fibroblástico 23 (FGF23)1–4.
En cuanto al manejo renal, en condiciones normales, la mayoría del fosfato filtrado se reabsorbe en el túbulo proximal, excretándose un 10-20% en la orina. La reabsorción en el túbulo proximal se realiza por 3 proteínas de transporte diferentes, dependientes del transporte de sodio concomitante1–4.
Aun siendo relevantes la absorción intestinal y la resorción ósea, es la reabsorción tubular renal la determinante de la fosfatemia. La reabsorción renal de fosfato depende de diversos factores, entre los que dominan la ingestión dietética de fosfato, la propia concentración de fosfatos en sangre y la actividad de las hormonas implicadas en su control: PTH, FGF23, otras fosfatoninas y la vitamina D.
Hormonas implicadas (fig. 1)1–4:
- •
PTH: su principal estímulo es la hipocalcemia en el receptor sensor de calcio paratiroideo, causando eliminación renal de fósforo. Así, tras inducir una mayor acción osteoclástica en el hueso que libera calcio y fósforo provenientes de la hidroxiapatita ósea, se consigue elevar las concentraciones de calcio, pero no de fosfato en sangre. Además, la PTH activa la 1-α-hidroxilasa para la síntesis de vitamina D activa o calcitriol.
- •
Vitamina D: para generar su forma activa, el precursor hormonal de producción cutánea (vitamina D3, 80%) o procedente de la ingestión (vitamina D2 o D3, 20%) se activa con 2 hidroxilaciones consecutivas: la hepática, en posición 25 (poco regulada), y la renal, en posición 1-α, esta última estimulada por la hipocalcemia y la hipofosfatemia y por las hormonas reguladoras PTH, calcitonina, hormona de crecimiento y factor de crecimiento insulínico 1, e inhibida por FGF23, por su producto, el calcitriol, y por la hiperfosfatemia. La vitamina D además aumenta la absorción de calcio y de fósforo ?este por mecanismos desconocidos? y puede modular acciones tanto activadoras como inhibidoras de la resorción ósea, quizá por un aumento del ligando del receptor activador para el factor nuclear κ B ?RANKL?. En el riñón aumenta la reabsorción de calcio tubular y estimula la producción de FGF23.
- •
FGF23: el FGF23-Klotho es una hormona peptídica producida por osteocitos, osteoblastos y células de estirpe mesenquimal, que se estimula por concentraciones elevadas de fósforo y calcitriol. Precisa para conformar su receptor al cofactor Klotho. Causa fosfaturia por inhibir su reabsorción tubular al evitar la expresión de los trasportadores, a la vez que inhibe la 1-α-hidroxilasa y activa la 24-hidroxilasa, reduciendo las concentraciones de calcitriol y la absorción de fosfato (y calcio).

A. Tejidos y hormonas implicados en la regulación del metabolismo del fosfato. B. Respuesta integrada para corregir la hipocalcemia. Adaptada de Fukumoto4.
Respecto a la integración de la acción hormonal con la ingestión dietética, tanto la baja ingestión de fosfato como la hipofosfatemia aumentan la reabsorción renal de fosfato. A diferencia de lo que ocurre con el calcio, en mamíferos no está caracterizado un receptor sensor de fosfato, pero tanto la PTH como el FGF 23 ?principales hormonas fosfatúricas? actúan aumentando la excreción de fosfatos. El mecanismo por el que una elevada ingestión dietética de fosfatos o la hiperfosfatemia promueven la liberación de FGF23 y de hormona D activa es desconocido en la actualidad2–4.
El metabolismo del fosfato está finamente regulado desde las paratiroides, el riñón o el hueso, pero con un papel clave del riñón en la modulación de sus concentraciones y el FGF23 emerge como la principal hormona fosfatúrica. El déficit de FGF23 causa hiperfosfatemia, exceso de calcitriol y calcificaciones de los tejidos blandos, mientras que su exceso causa hipofosfatemia, hipofunción del sistema de la vitamina D y alteraciones del crecimiento1–4.
Etiología de la hipofosfatemiaLa hipofosfatemia se define como la concentración de fósforo sérico inferior a 2,5mg/dl, que puede ser leve (2,0-2,5mg/dl), moderada (1,0-2,0mg/dl) o grave (<1,0mg/dl). Las principales causas de hipofosfatemia se catalogan según el mecanismo fisiopatológico4–10 (tabla 1). A su vez, puede clasificarse según se trate de una hipofosfatemia aguda o crónica8,11 (tabla 2). La hipofosfatemia aguda suele ser debida a fenómenos de redistribución del fósforo y la crónica, a alteración de su reabsorción tubular.
Mecanismos y clasificación etiopatogénica de la hipofosfatemia
| Disminución de la absorción intestinal |
| Restricción dietética de fósforo severa, desnutrición, anorexia |
| Déficit de vitamina D o resistencia a su acción |
| Antiácidos o quelantes del fósforo |
| Malabsorción (resección intestinal, enfermedades de la mucosa, diarrea crónica, esteatorrea) |
| Alcoholismo |
| Aumento de pérdidas renales |
| Hiperparatiroidismo primario o secundario |
| Déficit de vitamina D |
| Enfermedades tubulares renales |
| Raquitismo hipofosfatémico hereditario con hipercalciuria |
| Síndrome de Fanconi, enfermedad de Dent y otras tubulopatías |
| Intolerancia a la fructosa |
| Aumento de factores fosfatúricos (FGF23, MEPE y otros) |
| Raquitismo hipofosfatémico ligado al cromosoma X |
| Raquitismo hipofosfatémico autosómico dominante |
| Raquitismo hipofosfatémico autosómico recesivo |
| Osteomalacia oncogénica |
| Cetoacidosis diabética |
| Acidosis metabólica y respiratoria |
| Expansión de volumen |
| Fármacos (calcitonina, diuréticos, esteroides, bicarbonato, quimioterápicos, antirretrovirales, aminoglucósidos, anticonvulsivantes, hierro parenteral) |
| Trasplante renal |
| Redistribución el espacio intracelular |
| Alcalosis respiratoria aguda |
| Crisis blástica leucémica |
| Recuperación de la acidosis metabólica |
| Recuperación de la hipotermia |
| Síndrome de realimentación |
| Hiperventilación (sepsis, intoxicación por salicilatos, alcoholismo, pánico, encefalopatía hepática) |
| Administración de azúcares (glucosa, fructosa) |
| Administración de insulina |
| Síndrome de hueso hambriento |
| Catecolaminas |
FGF23: factor de crecimiento fibroblástico 23; MEPE: fosfoglucoproteína de matriz extracelular.
Causas de hipofosfatemia
| Hipofosfatemia aguda |
| Aumento de pérdidas renales |
| Posterior a trasplante renal (primeras semanas) |
| Expansión de volumen aguda |
| Redistribución del espacio intracelular |
| Posterior a trasplante de médula ósea |
| Leucemia aguda y linfoma |
| Corrección de cetoacidosis diabética |
| Diuresis osmótica |
| Síndrome de realimentación |
| Alcalosis respiratoria aguda |
| Síndrome de hueso hambriento |
| Miscelánea |
| Sepsis severa |
| Grandes quemados |
| Diálisis inadecuada |
| Sobredosificación aguda de paracetamol |
| Intoxicación por salicilatos |
| Hipotermia |
| Hipofosfatemia crónica |
| Déficit de aporte |
| Nutrición parenteral inadecuada |
| Déficits nutricionales |
| Disminución de la absorción intestinal |
| Deficiencia de vitamina D |
| Raquitismo dependiente de la vitamina D |
| Tratamiento crónico con antiácidos |
| Síndromes malabsortivos |
| Enfermedad renal crónica |
| Alcoholismo crónico |
| Aumento de pérdidas renales por enfermedad renal primaria |
| Síndrome de Fanconi |
| Enfermedad de Dent |
| Tóxicos (ifosfamida, sales de platino, diuréticos, glucocorticoides, antirretrovirales, acetazolamida, aminoglucósidos) |
| Hiperparatiroidismo primario o secundario |
| Aumento de los niveles séricos de FGF23 |
| Raquitismos hipofosfatémicos |
| Osteomalacia tumoral |
| Displasia fibrosa |
| Síndrome de McCune-Albright |
| Tóxicos (hierro parenteral) |
| Redistribución del espacio intracelular |
| Tratamiento con agentes estimulantes de la eritropoyesis en pacientes con cirrosis |
FGF23: factor de crecimiento fibroblástico 23.
Adaptada de Bacchetta y Salusky11.
Fisiopatológicamente se pueden considerar 3 causas de hipofosfatemia:
- a.
Disminución de la absorción intestinal de fósforo: es excepcional, debido a la ubicuidad del fosfato en los alimentos.
- b.
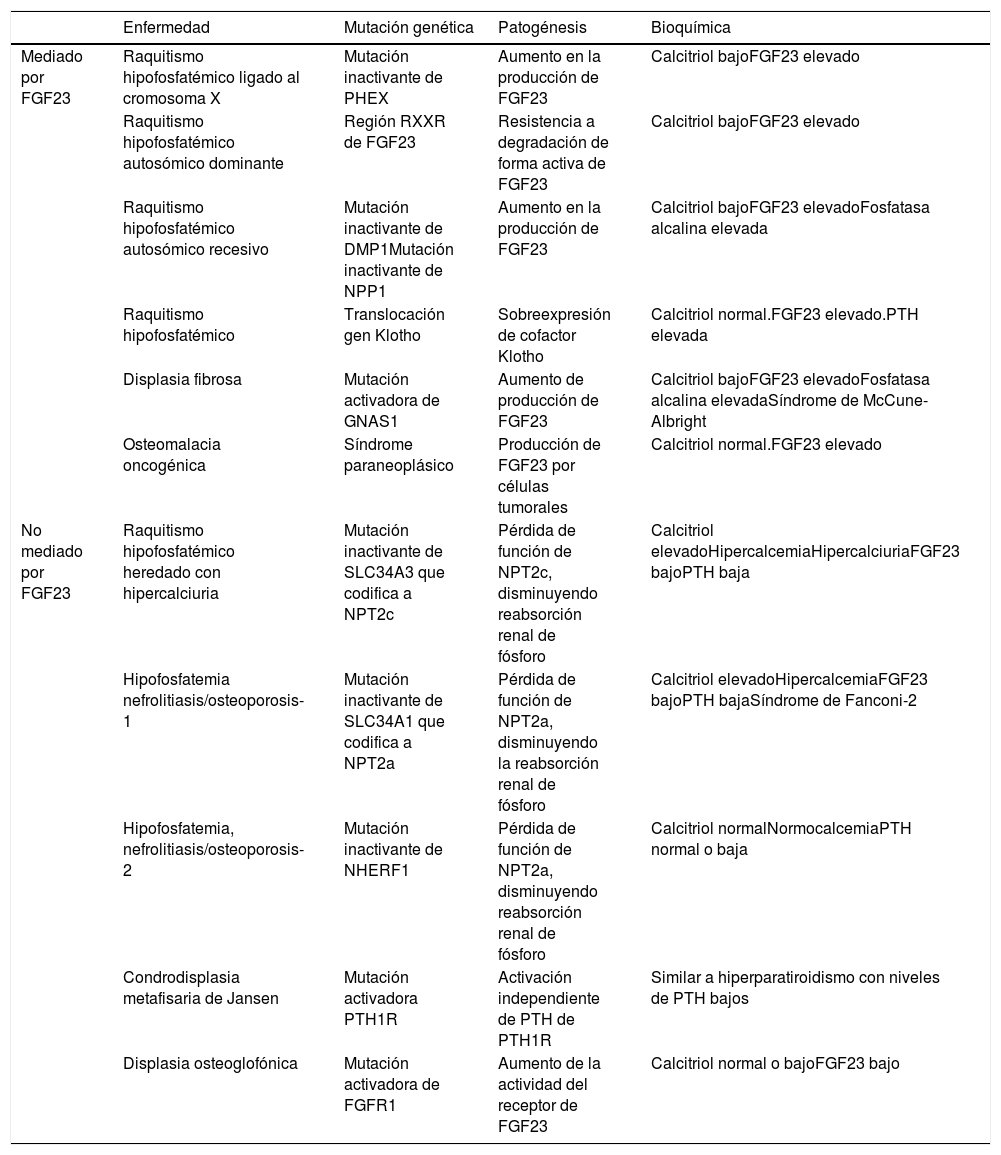
Aumento de las pérdidas renales de fósforo: es la causa más común de hipofosfatemia. Puede ser o no ser mediado por FGF23 (tabla 3), de origen genético, tumoral o adquirido12–14.Otras causas son el tratamiento con diuréticos como acetazolamida, tiazidas, diuréticos de asa y manitol, así como el hiperparatiroidismo primario hiper y normocalcémico y el hiperparatiroidismo secundario asociado a función renal normal con hipocalcemia.
Tabla 3.Causas de hipofosfatemia por aumento de la excreción renal de fosfato
Enfermedad Mutación genética Patogénesis Bioquímica Mediado por FGF23 Raquitismo hipofosfatémico ligado al cromosoma X Mutación inactivante de PHEX Aumento en la producción de FGF23 Calcitriol bajoFGF23 elevado Raquitismo hipofosfatémico autosómico dominante Región RXXR de FGF23 Resistencia a degradación de forma activa de FGF23 Calcitriol bajoFGF23 elevado Raquitismo hipofosfatémico autosómico recesivo Mutación inactivante de DMP1Mutación inactivante de NPP1 Aumento en la producción de FGF23 Calcitriol bajoFGF23 elevadoFosfatasa alcalina elevada Raquitismo hipofosfatémico Translocación gen Klotho Sobreexpresión de cofactor Klotho Calcitriol normal.FGF23 elevado.PTH elevada Displasia fibrosa Mutación activadora de GNAS1 Aumento de producción de FGF23 Calcitriol bajoFGF23 elevadoFosfatasa alcalina elevadaSíndrome de McCune-Albright Osteomalacia oncogénica Síndrome paraneoplásico Producción de FGF23 por células tumorales Calcitriol normal.FGF23 elevado No mediado por FGF23 Raquitismo hipofosfatémico heredado con hipercalciuria Mutación inactivante de SLC34A3 que codifica a NPT2c Pérdida de función de NPT2c, disminuyendo reabsorción renal de fósforo Calcitriol elevadoHipercalcemiaHipercalciuriaFGF23 bajoPTH baja Hipofosfatemia nefrolitiasis/osteoporosis-1 Mutación inactivante de SLC34A1 que codifica a NPT2a Pérdida de función de NPT2a, disminuyendo la reabsorción renal de fósforo Calcitriol elevadoHipercalcemiaFGF23 bajoPTH bajaSíndrome de Fanconi-2 Hipofosfatemia, nefrolitiasis/osteoporosis-2 Mutación inactivante de NHERF1 Pérdida de función de NPT2a, disminuyendo reabsorción renal de fósforo Calcitriol normalNormocalcemiaPTH normal o baja Condrodisplasia metafisaria de Jansen Mutación activadora PTH1R Activación independiente de PTH de PTH1R Similar a hiperparatiroidismo con niveles de PTH bajos Displasia osteoglofónica Mutación activadora de FGFR1 Aumento de la actividad del receptor de FGF23 Calcitriol normal o bajoFGF23 bajo DMP1: fosfoproteína ácida de la matriz de la dentina 1; FGF23: factor de crecimiento fibroblástico 23; FGFR1: receptor 1 factor de crecimiento fibroblástico; NPP1: nucleótido pirofosfatasa/fosfodiesterasa 1; NPT2c: trasportador renal de fosfato dependiente de sodio tipo 2c; NPT2a: trasportador renal de fosfato dependiente de sodio tipo 2c; PTH: parathormona; PTH1R: receptor 1 hormona paratiroidea.
- c.
Desplazamiento del fósforo del espacio extracelular al intracelular: diversas situaciones o tratamientos pueden provocar un estímulo de la glucólisis con aumento de la captación celular de fosfato y/o consumo de fosfato por aumento del anabolismo.
Los signos y síntomas de la hipofosfatemia se producen cuando esta va acompañada de una depleción del fosfato intracelular (no así cuando la hipofosfatemia es debida únicamente a un movimiento de fosfato hacia la célula). Así, la hipofosfatemia aguda grave ocurre principalmente en pacientes hospitalizados con procesos médicos y quirúrgicos graves, que presentan un déficit agudo y una depleción preexistente de fosfato; la hipofosfatemia aguda sin agotamiento crónico previo de fosfato no suele ser sintomática15.
1. Efectos en el metabolismo mineral:
- –
A nivel renal: inhibe la reabsorción tubular de calcio y magnesio, produciendo hipercalciuria16.
- –
A nivel óseo aumenta la resorción ósea con un incremento de la calcemia que contribuye a la hipercalciuria. De forma prolongada puede dar lugar a raquitismo y osteomalacia17.
2. Efectos sobre otros sistemas por déficit de adenosín trifosfato y 2-3 difosfoglicerato eritrocitario:
El descenso de 2-3 difosfoglicerato eritrocitario aumenta la afinidad de la hemoglobina por el oxígeno y reduce la liberación de oxígeno a nivel tisular. El descenso de adenosín trifosfato produce alteración en las funciones celulares que dependen de este para la obtención de energía. Estas alteraciones se traducen en diversos síntomas a distintos niveles:
- –
Sistema nervioso central: desde una leve irritabilidad y parestesia hasta manifestaciones más severas con encefalopatía metabólica, delirium, convulsiones generalizadas y coma18. También puede contribuir al desarrollo de mielinolisis central pontina19.
- –
Sistema cardiopulmonar: alteración de la contractilidad miocárdica, especialmente en pacientes con hipofosfatemia grave (<1mg/dl). También se ha asociado a arritmias ventriculares y a una mayor necesidad de fármacos vasoactivos en cirugía cardiaca20. Afecta también a la contractilidad diafragmática y, por tanto, la función pulmonar21.
- –
Músculo esquelético y músculo liso: miopatía proximal, disfagia e íleo. En el síndrome de realimentación y en alcohólicos se han descrito también episodios de rabdomiólisis22.
- –
Manifestaciones hematológicas: hemólisis y deterioro de la función leucocitaria (reducción de la fagocitosis y quimiotaxis granulocitaria). Son raras y se dan a concentraciones muy bajas de fósforo (<0,5mg/dl). También puede haber alteración plaquetaria con retracción defectuosa del coágulo y trombocitopenia15.
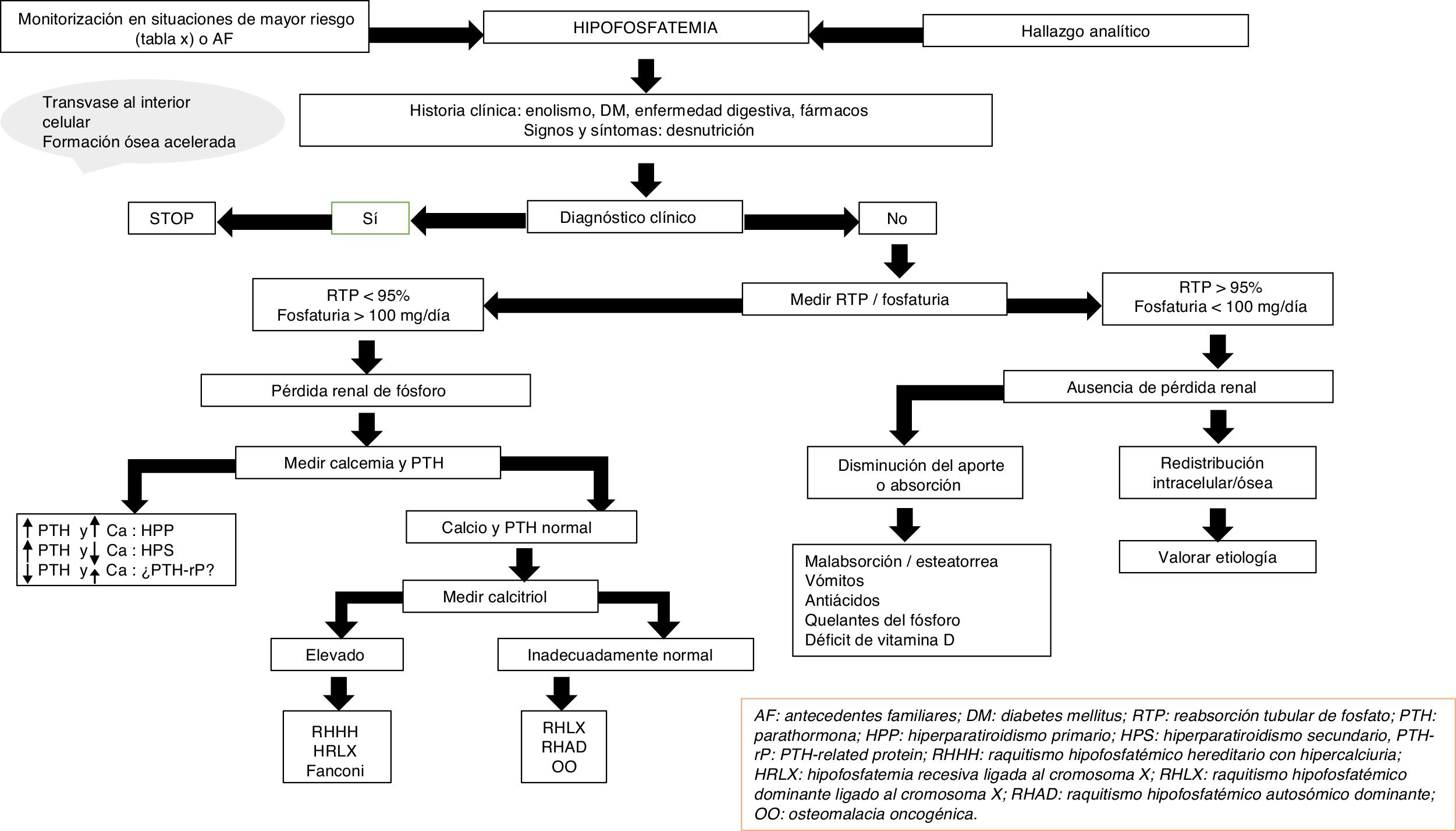
Con frecuencia, la etiología de la hipofosfatemia es evidente tras la realización de una adecuada historia clínica. En el resto de los casos, y en ausencia de seudohipofosfatemia causada por paraproteínas23, es necesario medir la excreción urinaria de fosfato mediante su determinación en orina de 24h o el cálculo de la reabsorción tubular de fósforo en una muestra aislada de orina. La respuesta fisiológica a nivel renal ante un descenso de los niveles séricos de fosfato es un aumento de la reabsorción de este, que conlleva prácticamente una abolición de la fosfaturia.
En este sentido, la presencia de niveles de fosfato inferiores a 100mg al día en la recogida de orina de 24h o una reabsorción tubular de fósforo superior al 95% orienta a una causa extrarrenal, entre la que podemos destacar la redistribución interna del fosfato o un descenso de la absorción intestinal del mismo15. La redistribución intracelular es más frecuente en pacientes que reciben perfusiones de glucosa o insulina, como acontece en el síndrome de realimentación o en el tratamiento de la hiperglucemia aguda en la diabetes mellitus. A su vez, también puede presentarse en pacientes con hiperventilación y alcalosis respiratoria aguda. A nivel intestinal, diversos mecanismos pueden conllevar una disminución de la absorción de fosfato, un aumento del tránsito y las secreciones en la diarrea crónica, la formación de sales insolubles en el tratamiento con suplementos de calcio, magnesio o aluminio y la inhibición del transporte intestinal en la terapia concomitante con niacina15.
Por otro lado, una fosfaturia superior o igual a 100mg al día o una reabsorción tubular de fósforo inferior al 95% indican una pérdida renal de fosfato y orientan a un exceso de hormonas o péptidos fosfatúricos (PTH, FGF23, Klotho), una alteración en el metabolismo de la vitamina D o, en menor medida, un defecto tubular primario. Tanto el hiperparatiroidismo primario como el secundario pueden asociarse a hipofosfatemia15. La determinación de calcio sérico y urinario, función renal, niveles de magnesio, vitamina D y el consumo de fármacos, entre otros, ayudarán al diagnóstico diferencial entre ambas entidades. La determinación de PTH-rp debe realizarse en situaciones de hipercalcemia con niveles bajos de PTH intacta.
En ausencia de hiperparatiroidismo y alteración en los niveles de calcio debemos pensar en la presencia de formas genéticas de raquitismo hipofosfatémico o en el síndrome paraneoplásico de osteomalacia oncogénica (OO)24,25. La edad de inicio, el perfil clínico, los niveles de calcitriol y FGF23, y la solicitud de pruebas genéticas nos ayudarán al diagnóstico diferencial entre las distintas entidades (fig. 2).

El raquitismo hipofosfatémico ligado a X (RHLX), el raquitismo hipofosfatémico autosómico dominante (RHAD) y la OO cursan con niveles disminuidos o inapropiadamente normales de calcitriol y, en general, con cifras elevadas de FGF2325. Los niveles elevados de esta hormona conllevan una disminución de la reabsorción renal de fosfato y de la síntesis compensatoria de 1,25-dihidroxivitamina D. En niños, el análisis genético del gen PHEX ayuda al diagnóstico de RHLX, si bien la ausencia de mutaciones conocidas no descarta la enfermedad, ya que se ha observado que solo están presentes en el 50-70% de los pacientes26. Se debe establecer el diagnóstico diferencial con el raquitismo hipofosfatémico autosómico recesivo, que es una entidad extremadamente rara que cursa con hallazgos similares al RHLX y se suele presentar en la infancia tardía. El análisis de los genes responsables de la síntesis de proteínas relacionadas con la degradación de FGF23 establece el diagnóstico de los 3 subtipos conocidos hasta la fecha27,28.
En adultos, la constatación de niveles previos normales de fosfato en sangre demuestra la presencia de OO, si bien, en ocasiones, los pacientes con RHAD también pueden cursar con cifras normales25. La OO es ocasionada por tumores mesenquimales con secreción ectópica de FGF23 y otras fosfatoninas. Con cierta frecuencia la localización del tumor responsable es un reto diagnóstico en el que se han empleado diferentes técnicas radiológicas y nucleares. Recientemente se ha propuesto como aproximación diagnóstica la realización escalonada de tomografía por emisión de positrones-tomografía computarizada con péptidos análogos de la somatostatina, como DOTA0-Tyr3-octreotate marcado con galio 68, muestreo venoso sistémico de FGF23 y resonancia magnética nuclear de 3 teslas29. Hasta que este esquema diagnóstico sea una realidad en la práctica clínica habitual, el abordaje inicial debe seguir realizándose con las técnicas más habituales (Octreoscan, tomografía por emisión de positrones-tomografía computarizada con fluorodesoxiglucosa, tomografía computarizada o resonancia magnética nuclear). Aunque el RHAD es muy poco frecuente, la ausencia de OO obliga al estudio genético de la misma. Diferentes mutaciones en el gen de FGF23 se han asociado a una resistencia de esta a su degradación proteolítica. Sin embargo, sus niveles no están consistentemente elevados, lo que se ha achacado a la presencia de periodos quiescentes en los que los niveles de fósforo y FGF23 son normales en este grupo de pacientes30. A su vez, estudios más recientes han asociado el déficit de hierro con niveles elevados de FGF23 en pacientes con RHAD31.
Por último, varios defectos tubulares se han relacionado con un incremento de la pérdida renal de fosfato15,25. Además de con hipofosfatemia cursan con hipercalciuria que puede causar nefrocalcinosis, litiasis y fallo renal, así como otras alteraciones específicas de cada uno de los síndromes. El raquitismo hipofosfatémico hereditario con hipercalciuria es un trastorno genético caracterizado por la pérdida de función de uno de los subtipos de cotransportadores de sodio y fosfato a nivel renal32. Cursa con niveles elevados de calcitriol por una respuesta conservada de la síntesis de este a los niveles descendidos de fosfato. La elevación de este y la presencia de hipercalciuria ayudan al diagnóstico junto con el análisis genético. En la hipofosfatemia recesiva ligada al cromosoma X, o enfermedad de Dent, las alteraciones bioquímicas son similares a las del raquitismo hipofosfatémico hereditario con hipercalciuria, si bien las mujeres portadoras presentan únicamente hipercalciuria25,33. El síndrome de Fanconi cursa con un defecto generalizado de la función del túbulo proximal que ocasiona otras alteraciones, como glucosuria, hipouricemia, aminoaciduria y acidosis metabólica por pérdida de bicarbonato. Al igual que el raquitismo hipofosfatémico hereditario con hipercalciuria, se han observado niveles elevados de calcitriol15.
Tratamiento de la hipofosfatemiaEl tratamiento de la hipofosfatemia depende de la causa y de otros factores como la cronicidad, la severidad, las manifestaciones clínicas, la presencia de hipercalcemia o hipocalcemia y la función renal. Los síntomas manifiestos de hipofosfatemia son raros, a menos que sus concentraciones séricas disminuyan por debajo de 2mg/ml. Los síntomas graves, como la debilidad muscular o la rabdomiólisis, suelen aparecer cuando las concentraciones de fosfato disminuyen por debajo de 1mg/dl7.
En la hipofosfatemia aguda con depleción de fosfato la suplementación de este puede realizarse por vía oral o intravenosa. La repleción por vía oral es más segura, pero su absorción es impredecible y puede provocar efectos adversos gastrointestinales, como diarrea. La repleción intravenosa corrige la hipofosfatemia más rápidamente, pero tiene el riesgo de provocar hipocalcemia, arritmias, calcificaciones ectópicas y fracaso renal agudo34. En la hipofosfatemia aguda severa (<1mg/dl) con depleción de fosfato normalmente se requiere el tratamiento intravenoso, particularmente en pacientes ingresados en la unidad de cuidados intensivos. En estos casos pueden administrarse dosis de entre 0,25 y 0,50mmol/kg de fosfato sódico o potásico durante 8-12h hasta un máximo de 80mmol. Es necesario monitorizar estrictamente los niveles de calcio y fósforo para reducir el riesgo de calcificaciones ectópicas y otras complicaciones35. En casos menos graves el tratamiento puede realizarse con suplementos orales de fósforo de entre 1-4g por día.
La hipofosfatemia crónica es consecuencia de pérdidas gastrointestinales o renales de fosfato. En ocasiones la corrección de la causa de hipofosfatemia requiere la retirada de los quelantes de fosfato, los diuréticos o la corrección de la hipomagnesemia. En casos leves, el aumento de la ingesta dietética de fósforo (medio litro de leche desnatada de vaca aporta unos 450mg de fósforo) puede ser suficiente. Las pérdidas renales pueden deberse a enfermedades genéticas caracterizadas por un aumento de la actividad del FGF23, como en el RHLX o la OO36. En estos casos la terapia con fosfato oral está indicada para corregir las anormalidades óseas y restablecer el crecimiento normal en niños. El tratamiento convencional de los trastornos que implican a FGF23 incluye dosis altas de suplementos de fosfato (2-4g por día) en dosis fraccionadas y calcitriol (0,25-2μg/día)37. En la OO el objetivo es la extirpación del tumor responsable.
Recientemente se ha desarrollado un anticuerpo monoclonal frente a FGF23 denominado burosumab, que ha demostrado su eficacia en diferentes formas de hipofosfatemia genéticas o adquiridas. El tratamiento con burosumab a dosis de hasta 1mg/kg cada 4 semanas aumenta la reabsorción tubular de fosfato, normaliza las concentraciones de fósforo e incrementa las concentraciones de calcitriol38.
HiperfosfatemiaSe define como concentración de fósforo sérico superior a 4,5mg/dl (1,45mmol/l) en los adultos, y superior a 7mg/dl (2,26mmol/l) en los niños, confirmada en 2 determinaciones.
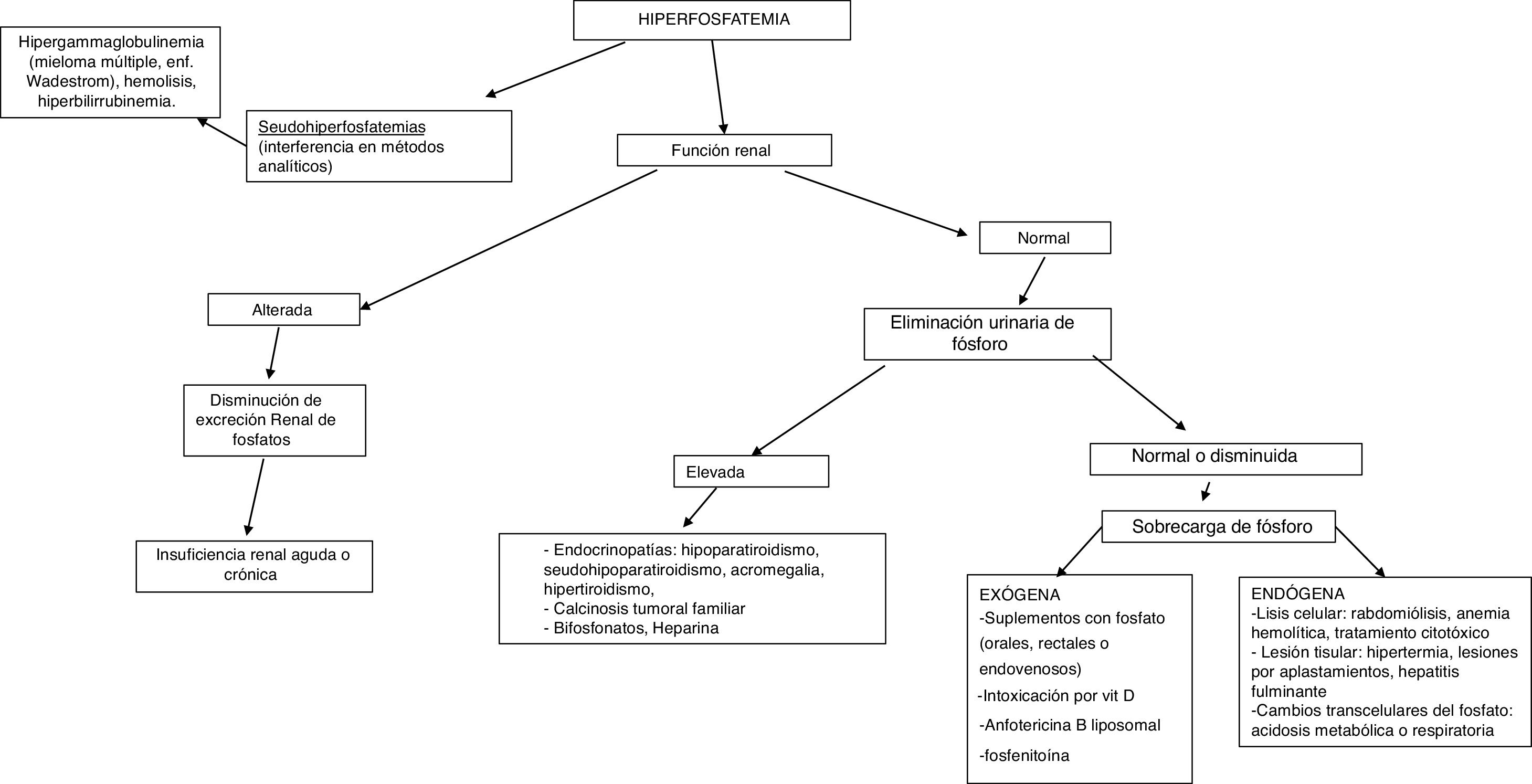
La causa más frecuente de hiperfosfatemia es la disminución en la excreción renal de fosfato (tabla 4), principalmente por insuficiencia renal aguda o enfermedad renal crónica (ERC) cuando existe un filtrado glomerular inferior a 20ml/min/1,73m2. En ocasiones, puede estar ocasionada por un aumento en su reabsorción tubular, redistribución desde el compartimento intracelular al extracelular o incremento en su aporte. La hiperfosfatemia espuria o seudohiperfosfatemia se debe a la interferencia con los métodos analíticos: hiperglobulinemia, hiperlipidemia, hemólisis e hiperbilirrubinemia, tratamiento con altas dosis de anfotericina B liposomal, contaminación de la muestra con activador del plasminógeno tisular recombinante o heparina.
Causas de hiperfosfatemia
| Excreción alterada de fosfato renal |
| Insuficiencia renal crónica (filtrado glomerular<20ml/min/1,73m2) |
| Descenso de FGF23: calcinosis tumoral familiar y no familiar |
| Endocrinopatías: acromegalia, hipoparatiroidismo y seudohipoparatiroidismo |
| Déficit de magnesio |
| Síndrome de leche y alcalinos |
| Fármacos: heparina, bifosfonatos |
| Incremento del fosfato intracelular |
| A. Aumento del aporte (intravenoso, oral, rectal) |
| Sales de fosfato (laxantes orales o rectales) |
| Fosfenitoína |
| Anfotericina B liposómica |
| B. Cambios transcelulares del fosfato: acidosis metabólica o respiratoria |
| C. Catabolismo o lisis celular rápida |
| Estados catabólicos |
| Lesión tisular: hipertermia, lesiones de aplastamiento, hepatitis fulminante |
| Lisis celular: anemia hemolítica, rabdomiólisis, tratamiento citotóxico, leucemia grave |
Habitualmente la hiperfosfatemia es leve y asintomática; sin embargo, la hiperfosfatemia crónica es un factor importante en el desarrollo del hiperparatiroidismo secundario en la ERC. En la hiperfosfatemia aguda severa, las manifestaciones clínicas derivan de la hipocalcemia causada por la formación de sales de fosfato cálcico insolubles: debilidad a nivel musculoesquelético, tetania e incremento de la excitabilidad neuromuscular; a nivel del sistema nervioso central puede provocar crisis comiciales y deterioro cognitivo.
Las manifestaciones clínicas de la hiperfosfatemia crónica están relacionadas con la localización de las calcificaciones ectópicas de los tejidos blandos: prurito, rotura tendinosa, queratopatía en banda y calcificaciones vasculares. Estas pueden localizarse en pequeñas arteriolas y capilares (calcifilaxis), produciendo lesiones cutáneas necróticas y hemorragias subungueales en las arterias de mediano calibre; pueden dar lugar a un síndrome coronario agudo y arritmias cardíacas, prolongación del intervalo QT o valvulopatías3,39.
La hiperfosfatemia crónica se ha asociado con un aumento de la mortalidad en pacientes con ERC en prediálisis: existe un 35% de aumento de mortalidad por cada miligramo de incremento de fosfatemia por encima de los valores de normalidad; sin embargo, otros estudios no han demostrado esta asociación. En pacientes con ERC en diálisis, el riesgo de mortalidad aumenta un 18% por cada miligramo de incremento por encima de los valores normales40.
Para el diagnóstico etiológico se recomienda la determinación sérica de 25-hidroxivitamina D, calcitriol, PTH, calcio corregido por albúmina, magnesio, creatinina, urea, fosfatasa alcalina, pH sérico y medición urinaria de creatinina, calcio y fósforo3. Una excreción fraccional de fósforo en orina inferior al 5% indica origen renal y si es superior señala un exceso de aporte (fig. 3).

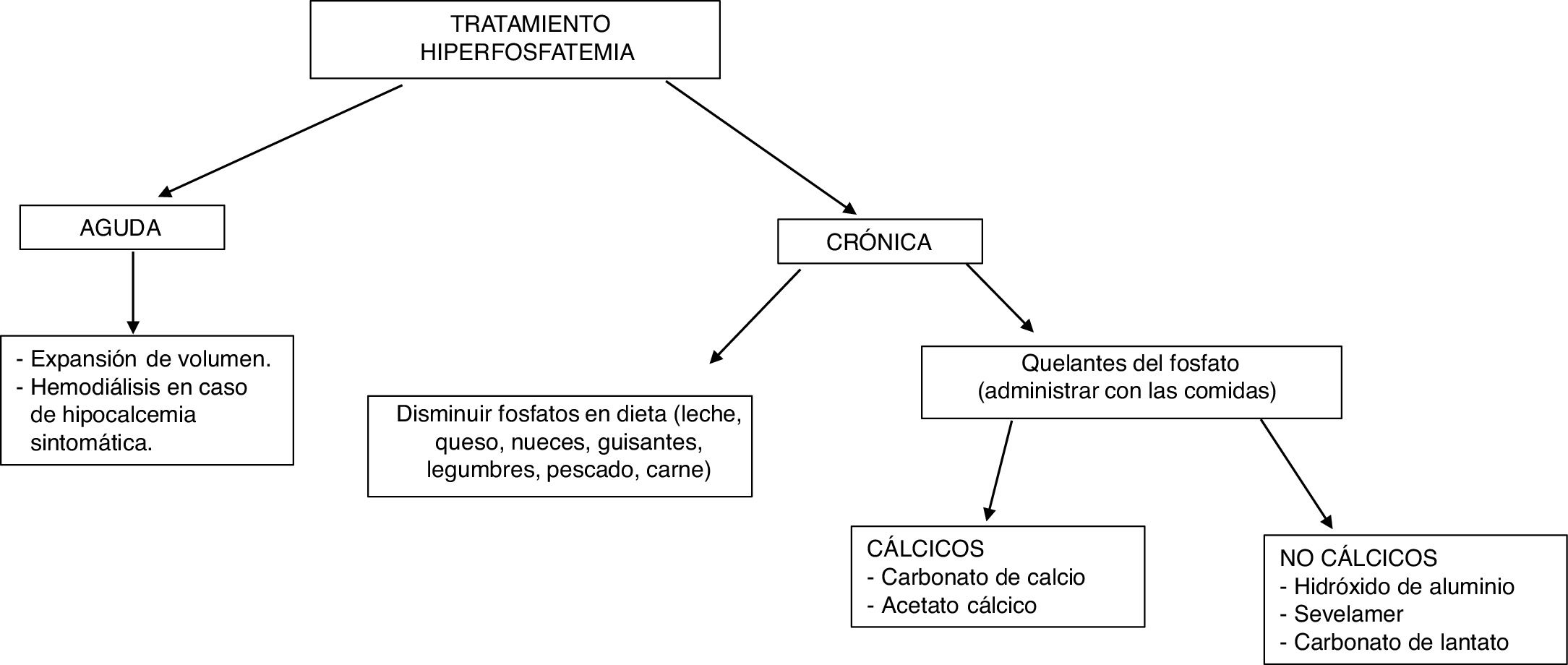
El tratamiento de la hiperfosfatemia va a depender de su causa, de la rapidez de instauración y de la presencia o no de insuficiencia renal (fig. 4).

Hiperfosfatemia aguda3,39. Las opciones terapéuticas son limitadas: la expansión de volumen puede ser útil si la función renal está conservada. Es fundamental identificar y suspender cualquier fuente exógena de fosfato; los antiácidos de hidróxido de aluminio reducen su absorción intestinal y favorecen la quelación del fosfato segregado por el intestino. La hemodiálisis es el tratamiento más eficaz de la hiperfosfatemia y se debe considerar en casos graves de instauración aguda.
Hiperfosfatemia crónica. En pacientes con ERC prediálisis, está indicado el tratamiento de la hiperfosfatemia progresiva o persistente40. Se recomienda restringir la ingesta dietética de fósforo (900mg/día) y usar quelantes si persiste la hiperfosfatemia; se puede iniciar tratamiento combinado en aquellos pacientes con niveles muy elevados (>6mg/dl). Los quelantes del fósforo producen un discreto descenso de la fosfatemia y en orina de 24h41. El descenso de las concentraciones de fósforo en estos pacientes no ha demostrado mejorar parámetros clínicos importantes como mortalidad o progresión de la ERC.
En pacientes con ERC en diálisis se recomienda restringir la ingesta de fósforo a 900mg/día asociada o no a quelantes del fósforo40. La restricción de ingesta de fósforo no ha demostrado aumentar la supervivencia de estos pacientes42. El uso de quelantes se ha asociado con un descenso del 25-29% de la mortalidad total43,44 y un 22% de mortalidad por causa cardiovascular14 tras un máximo de 3 años de seguimiento.
Como quelantes disponibles se encuentran el carbonato y el acetato cálcico (quelantes cálcicos), el sevelamer y el lantano (quelantes no cálcicos). El uso de quelantes no cálcicos se asocia con una reducción del 22-44% de la mortalidad comparado con el uso de quelantes cálcicos45,46. Este segundo grupo se ha asociado con hipercalcemia, enfermedad ósea adinámica y calcificaciones vasculares41. La elección del quelante del fósforo debe ser individualizada: se debe tener en cuenta la situación clínica del paciente, el coste del tratamiento, la tolerabilidad individual y otros parámetros del metabolismo mineral, como la PTH y la calcemia40.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interese.