




El gen CDKAL1 (proteína 1 asociada con la subunidad reguladora CDK5 1) codifica el gen proten1 asociado con la subunidad 5 dependiente de la quinasa 5 dependiente de ciclina (CDK5). Esta proteína ha demostrado contribuir a la regulación dependiente de glucosa de la secreción de insulina en islotes pancreáticos.
ObjetivosEl objetivo de nuestro estudio fue analizar los efectos de la variante genética rs7756992 del gen CDKAL1 sobre la glucosa en ayunas y la resistencia a la insulina, tras la pérdida de peso secundaria a la dieta hipocalórica con reemplazo parcial de comidas (pMRHD).
MétodosEl diseño fue no aleatorizado de un solo brazo de tratamiento con una dieta de fórmula en 44 sujetos con obesidad. Los pacientes recibieron educación nutricional y una dieta modificada con dos ingestas con una fórmula hiperproteica normocalórica durante tres meses. Los parámetros antropométricos y el perfil bioquímico se midieron en el tiempo basal y después de tres meses. Se evaluó la variante del gen CDKAL1 rs7756992.
ResultadosSe observó la siguiente distribución genética; (27AA [61,3%], 12 AG [27,3%] y 5 GG [11,4%]). Después de la pMRHD, el peso corporal, el índice de masa corporal (IMC), la masa grasa, la circunferencia de la cintura y la presión arterial disminuyeron en ambos genotipos. Los no portadores del alelo G mostraron una disminución significativa en los niveles de glucosa en ayunas (AA vs. AG + GG) (-6,1 ± 1,4 mg/dL vs. -1,2 ± 0,7 mg/dL; p = 0,01), niveles de insulina en ayunas (-3,6 ± 0,2 mU/L vs. -1,3 ± 0,6 mU/L; p = 0,02) y HOMA-IR (-1,2 ± 0,2 unidades vs. -0,3 ± 0,2 unidades; p = 0,01). Los niveles de glucosa en ayunas fueron más altos en los portadores del alelo G que en los no portadores del alelo G.
ConclusionesNuestros datos sugieren que la variante genética (rs7756992) del gen CDKAL1 está asociada con el estado glucémico después de una pMRHD, con una mejoría significativa de la glucosa en ayunas, la insulina y el HOMA-IR en sujetos sin alelo G.
The CDKAL1 (CDK5 Regulatory Subunit Associated Protein 1 Like 1) gene encodes cyclin-dependent kinase 5 (CDK5) regulatory subunit-associated proten1 like 1. This protein has been shown to contribute to the glucose-dependent regulation of insulin secretion in pancreatic islets.
AimsThe aim of our study was to analyze the effects of the rs7756992 genetic variant of CDKAL1 gene on fasting glucose and insulin resistance after weight loss secondary to partial meal replacement hypocaloric diet (pMRHD).
MethodsThis was a non-randomized, single-treatment study with a formula-diet in 44 obese subjects. The patients received nutritional education and a modified diet with two intakes of a normocaloric hyperproteic formula for 3-months. Anthropometric parameter and biochemical profile were measured at basal time and after 3 months. The variant of CDKAL1 gene rs7756992 was assessed.
ResultsThe following genetic distribution was observed; [27AA (61.3%), 12 AG (27.3%) and 5 GG (11.4%)]. After the pMRHD, body weight, the body mass index (BMI), fat mass, waist circumference and blood pressure decreased in both genotypes. Non-G allele carriers showed a significant improvement in fasting glucose levels (AA vs. AG + GG) (-6.1 ± 1.4 md/dL vs. -1.2 ± 0.7 mg/dl; p = 0.01), fasting insulin levels (-3.6 ± 0.2 mU/L vs. -1.3 ± 0.6 mU/L; p = 0.02) and HOMA-IR (-1.2 ± 0.2 units vs. -0.3 ± 0.2 units; p = 0.01). Fasting plasma glucose levels were higher in G allele carriers than non G allele carriers.
ConclusionsOur data suggest that the genetic variant (rs7756992) of CDKAL1 gene is associated with glycaemic status after a pMRHD, with greater improvements in fasting glucose, insulin and HOMA-IR in subjects without the G allele.
La obesidad es una pandemia en crecimiento, por otra parte, la diabetes mellitus tipo 2 (DM2) se ha entendido tradicionalmente como una consecuencia del síndrome metabólico inducido por la resistencia a la insulina secundaria a la obesidad. La prevalencia de obesidad en el mundo es del 12%1, mientras que en España es del 22%2. La piedra angular de todas las opciones de tratamientos para la obesidad incluye una dieta baja en calorías con ejercicio, con el objetivo de lograr una pérdida de peso clínicamente significativa de al menos un 5-10%. Incluso esta cantidad relativamente pequeña de pérdida de peso puede reducir el riesgo de desarrollar DM2, resistencia a la insulina y proteger la función de las células beta. Existe evidencia de que las dietas hipocalóricas con reemplazo parcial de comidas (pMRHD) son útiles para inducir la pérdida de peso. Un interesante metaanálisis3 que compara los programas de reemplazo parcial con las dietas tradicionales basadas en alimentos con restricción energética demostró que las pMRHD produjeron una pérdida del 7% en el peso corporal, en comparación con el 3% en las dietas tradicionales.
Existen vías metabólicas que relacionan la obesidad, la DM2 y el empeoramiento de la función de la célula beta. El gen CDKAL1 (proteína asociada a la subunidad reguladora CDK5 1 como 1) en el cromosoma 6p22.3 codifica la proteína asociada con la subunidad reguladora de la quinasa dependiente de ciclina 5 (CDK5)4. Esta proteína de 65 kD comparte homología de aminoácidos con CDK5RAP1, un inhibidor de la activación de CDK55. La CDK5 juega un papel principal en el desarrollo neuronal6, pero también tiene varias actividades extracerebrales7. Se ha demostrado que esta proteína contribuye a la regulación dependiente de la glucosa de la secreción de insulina en los islotes pancreáticos8. El mecanismo consiste en fosforilar el canal de Ca2+ dependiente de voltaje de tipo L en la Ser783, inhibiendo así la salida de Ca2+ hacia las células beta y la secreción de insulina9. Dado que la actividad excesiva de CDK5 está asociada con la pérdida de la función de las células beta y la reducción de la secreción de insulina en condiciones de glucosa alta, la regulación negativa de CDK5 mediada por CDKAL1 beneficiaría la función normal de las células beta. Finalmente, las variantes de CDKAL1 asociadas con esta disminución de la función de las células beta han mostrado una asociación significativa con DM2 en caucásicos10–11. Por ejemplo, el polimorfismo de un solo nucleótido (SNP) rs7756992 (c.371 + 30101 A > G) mostró un odds ratio (OR) de 1,09 para que los portadores del alelo G desarrollen DM212.
En sujetos con obesidad con DM2, un trabajo previo13 demostró que la variante genética rs7756992 del gen CDKAL1 está asociada con la respuesta del fármaco a un inhibidor de DPP-4. Sin embargo, no existen estudios en la literatura con intervención dietética con una pérdida de peso significativa que evalúen el efecto de este SNP en el metabolismo glucémico.
El objetivo de nuestro estudio fue analizar los efectos de la variante genética rs7756992 del gen CDKAL1 sobre la resistencia a la glucosa y la insulina en ayunas, después de una pérdida de peso significativa secundaria a pMRHD en sujetos con obesidad sin DM2.
MétodosDiseño del estudioDurante el período de reclutamiento de pacientes (de enero de 2018 a septiembre de 2019), se obtuvo el consentimiento informado por escrito de cada paciente elegible. Este estudio de intervención fue aprobado por nuestro Comité de Ética (Comité HCUVA 07/2018), en conformidad con la Declaración de Helsinki. Los pacientes fueron informados de su derecho a retirarse del estudio sin comprometer su tratamiento. El diseño fue de un estudio no aleatorizado, de tratamiento único con pMRHD con una fórmula normocalórica-hiperproteica. El reclutamiento de 44 pacientes con obesidad se realizó con un método consecutivo de muestreo entre los sujetos enviados a nuestra Unidad de Nutrición desde Atención Primaria. Los criterios de exclusión fueron cualquiera de los siguientes: antecedentes de enfermedad tiroidea, ataque cardíaco, ictus, trastornos renales o hepáticos graves, alcoholismo activo, tumor maligno, y que dentro de los tres meses anteriores al estudio estuvieran recibiendo medicamentos que influyen en los lípidos (terapia hormonal, glucocorticoides y medicamentos antiinflamatorios) o niveles de glucosa (metformina, sulfonilureas, tiazolidinediona, insulina, antagonistas del receptor GLP-1, S-GLT2, inhibidores de DPP-IV u otro medicamento relacionado con el tratamiento de la diabetes mellitus). Los sujetos con obesidad eran elegibles para participar si cumplían los criterios siguientes criterios de inclusion: índice de masa corporal (IMC) superior a 35 kg/m2 y la edad entre 30 y 70 años.
Después de obtener el consentimiento informado por escrito, recolectamos 10 mL de sangre venosa periférica en tubos que contienen EDTA-Na2. Después de etiquetar cada muestra con un número de estudio anonimizado, medimos los niveles de insulina, colesterol total, colesterol LDL, colesterol HDL y triglicéridos. La variante del gen CDKAL1 se evaluó por reacción en cadena de la polimerasa. Todos los parámetros antropométricos (peso, altura, IMC, circunferencia de la cintura [CC] y masa grasa por impedancia) y la presión arterial se registraron en el tiempo basal y también después de tres meses.
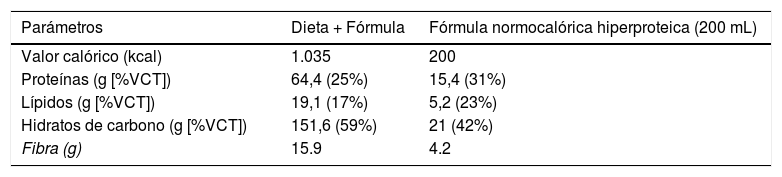
Intervención dietéticaDespués de la inclusión del paciente en el estudio, se aconsejó a todos los sujetos sobre las formas de modificar su dieta habitual a una dieta hipocalórica de reemplazo parcial de comidas (pMRHD), con dos bricks de una fórmula hiperproteica normocalórica. Las características nutricionales de la pMRHD y la fórmula artificial se muestran en la tabla 1. Esta intervención dietética se estructura en cinco comidas (desayuno, merienda, almuerzo, merienda, cena, merienda después de la cena). El almuerzo y la cena fueron reemplazados por la fórmula normocalórica-hiperproteica (Vegestart® Complet), cuyas características nutricionales se describen en la tabla 1. Un dietista realizó un refuerzo por teléfono cada siete días. Los diferentes parámetros se llevaron a cabo antes del inicio de la intervención dietética y a los tres meses después del comienzo de la intervención.
Composición dietética de la dieta de cinco ingestas (tres ingestas como alimento natural y dos ingestas como fórmula artificial), como se indica en la sección «Material y método»
| Parámetros | Dieta + Fórmula | Fórmula normocalórica hiperproteica (200 mL) |
|---|---|---|
| Valor calórico (kcal) | 1.035 | 200 |
| Proteínas (g [%VCT]) | 64,4 (25%) | 15,4 (31%) |
| Lípidos (g [%VCT]) | 19,1 (17%) | 5,2 (23%) |
| Hidratos de carbono (g [%VCT]) | 151,6 (59%) | 21 (42%) |
| Fibra (g) | 15.9 | 4.2 |
Fórmula normocalórica hiperproteica Vegestart® Complet.
%VCT: porcentaje del valor calórico total.
El peso se determinó sin ropa con una precisión de ± 100 g, utilizando una báscula automática (Seca, Birmingham, Reino Unido). La altura se determinó con el sujeto en posición vertical al centímetro más cercano, utilizando un estadiómetro manual (Seca Birmingham, Reino Unido). El IMC se calculó utilizando la ecuación habitual: peso (kg)/altura x altura (m2). Se calculó la diferencia de peso por el porcentaje de pérdida de peso (% PP): (peso antes de la intervención – peso después de la intervención [kg]/peso basal [kg]) x 100. La adiposidad abdominal se calculó por la circunferencia de la cintura (WC). La circunferencia de la cintura (CC) se midió en el diámetro más estrecho entre el proceso xifoides y la cresta ilíaca. Se realizó un análisis de impedancia bioeléctrica (BIA) en todas las materias. Se usó una corriente eléctrica alterna de 0,8 mA a 50 kHz, producida por un generador de señal calibrado (EFG, Akern, Firenze, Italia) y se aplicó a la piel mediante electrodos adhesivos ubicados en el dorso de la mano izquierda y el pie izquierdo14.
La presión arterial se midió tres veces después de un descanso de 10 min, con un esfigmomanómetro de mercurio cero al azar, y se promedió (Omron, LA, CA, EE. UU.).
Parámetros bioquímicosEl perfil lipídico general (niveles de colesterol total y triglicéridos) se determinó mediante una técnica colorimétrica enzimática (Technicon Instruments, Ltd., Nueva York, Nueva York, EE. UU.). El colesterol LDL se calculó utilizando la fórmula de Friedewald (colesterol LDL = colesterol total-colesterol HDL-triglicéridos/5)15. El colesterol HDL se determinó enzimáticamente en el sobrenadante después de la precipitación de otras lipoproteínas con sulfato de magnesio dextrano. La insulina se determinó mediante radioinmunoensayos (RIA Diagnostic Corporation, LA, CA), con una sensibilidad de 0,5 mUI/L (rango normal 0,5-30 mUI/L)16, los niveles de glucosa en plasma se determinaron mediante un método de glucosa oxidasa (analizador de glucosa 2, Beckman Instruments, Fullerton, CA, EE.UU.). La evaluación del modelo de homeostasis para la resistencia a la insulina (HOMA-IR) se obtuvo utilizando estos valores con esta ecuación: (insulina en ayunas [mU/L] x glucosa [mmol/L]/22,5)17.
Estudio genéticoEl genotipado (rs7756992) se realizó mediante la plataforma de genotipado TaqMan™ OpenArray™ (ThermoFisher, Pittsburgh, Pens, EE. UU.). Las muestras se cargaron usando el sistema AccuFill, y la amplificación se realizó en el instrumento QuantStudio 12K Flex Real-Time qPCR (ThermoFisher, Pittsburgh, Pens, EE. UU.). Un volumen total de 7,5 μl con 2,5 μl de TaqMan™ OpenArray™ Master Mix (Applied Biosystems, Foster City, CA, EE. UU.) y 2,5 μl de muestra de ADN humano se cargaron y amplificaron en matrices siguiendo las instrucciones del fabricante TaqMan Genotyper (LifeTechnologies, Carlsbad, CA, EE. UU.). La variante del gen CDKAL1 estaba en equilibrio Hardy-Weinberg (p = 0,33).
Dieta hipocalórica de reemplazo parcial de comidas (pMRHD)Antes y después de tres meses de la intervención, todos los sujetos con obesidad completaron un registro dietético de 72 h para evaluar la ingesta de calorías y nutrientes. El primer registro se realizó antes de comenzar la intervención para evaluar las ingestas dietéticas basales. El segundo registro dietético se realizó para evaluar la adherencia a pMRHD. Los registros fueron revisados por un dietista y analizados por el programa (Dietsource®, Nestlé, Ginebra, Suiza). Se utilizaron tablas de composición nacional de alimentos como referencia18. Las recomendaciones de ejercicio para pacientes de ambos grupos fueron actividades físicas aeróbicas, al menos cuatro veces por semana (una hora cada una). Los ejercicios prescritos a los sujetos fueron caminar, correr, andar en bicicleta, nadar y los ejercicios de fuerza muscular (entrenamiento con pesas o levantamiento de pesas) estaban contraindicados. La actividad de ejercicio de los pacientes fue autoinformada a través de un cuestionario autocompletado.
Análisis estadísticoLos datos se analizaron con el programa SPSS (versión 19.0 Armonk, NY, EE. UU.). Los resultados se muestran como media ± DE. El tamaño de la muestra se calculó para detectar diferencias en más de dos unidades en HOMA-IR con 90% de potencia y 5% de significación (n = 40). Se examinó la normalidad de cada variable con la prueba de Kolmogorov-Smirnov. Las variables categóricas se analizaron con la prueba de χ2, con la corrección de Yates, según sea necesario, y la prueba de Fisher. La importancia de la interacción entre las características clínicas/bioquímicas y el SNP se evaluó mediante un análisis de varianza (ANOVA), con prueba de Bonferroni post hoc. Para analizar el efecto de la intervención en cada grupo (p tiempo) y para analizar la interacción (p tiempo-grupo), que informa si el efecto de la intervención es el mismo para los dos grupos. También se utilizó la t de Student para el análisis de las diferencias entre los dos grupos en la variable principal (antes y después) (p genotipo). El análisis estadístico se realizó para los AG y GG combinados como grupo y el genotipo AA, como segundo grupo (genotipo de tipo salvaje), con un modelo dominante. La distribución del genotipo se probó para la desviación del equilibrio de Hardy-Weinberg mediante una prueba de χ2.
ResultadosSe incluyeron 44 sujetos con IMC > 35 kg/m2, con la siguiente distribución genética; [27AA (61,3%), 12 AG (27,3%) y 5 GG (11,4%)]. Todos los pacientes completaron el período de seguimiento de tres meses sin abandonos y no se informaron síntomas adversos relacionados con la intervención. La edad media fue de 61,1 ± 5,3 años (rango: 35-70 años) y el IMC medio 38,4 ± 2,1 kg/m2 (rango: 36,1-40,6 kg/m2). La edad fue similar en ambos grupos de genotipos (tipo salvaje [AA] vs. tipo mutante [AG + GG]) (62,0 ± 6,2 años vs. 60,9 ± 7,0 años: ns). En el grupo total, la distribución por sexo fue de 34 hombres (77,3%) y 10 mujeres (22,7%). La distribución por sexos fue similar en ambos grupos de genotipos, hombres (25,9 vs. 19,6%) y mujeres (74,1 vs. 80,4%).
La evaluación basal de la ingesta dietética (antes de que los sujetos con obesidad recibieran un pMRHD) con un registro de alimentos de tres días mostró los siguientes datos: ingesta total de calorías de 1.713,9 ± 491,81 cal/día, ingesta de carbohidratos de 160,2 + 53,9 g/día (39,6% de calorías), ingesta de grasas de 65,4 + 21,8 g/día (37% de calorías), ingesta de proteínas de 77,5 ± 20,1 g/día (23,4% de calorías) y consumo de fibra dietética 15,1 + 7,0 g/día. Después de los tres meses de pMRHD, estos sujetos alcanzaron las recomendaciones; 1.061,1 calorías por día, 148,1 ± 42,2 g/día de carbohidratos (64,2% de calorías), 27,9 ± 17,3 g/día de lípidos (23,2% de calorías), 62,3 ± 12,0 g/día de proteínas (23,6% de calorías) y 18,4 + 3,0 g/día de fibra dietética. La distribución de las grasas fue la siguiente; 32,7% de grasas saturadas, un 50,2% de grasas monoinsaturadas y un 17,1% de grasas poliinsaturadas. La actividad física fue similar en ambos grupos de genotipos (139,2 ± 21,3 min/semana frente a 141,0 ± 26,2 min/semana; p = 0,31).
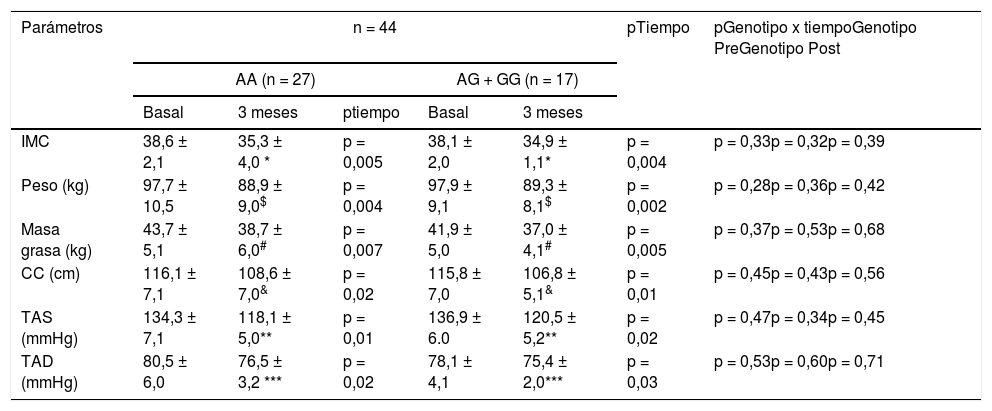
Como se nuestra en la tabla 2, no hubo diferencias estadísticas relacionadas con el genotipo (basal y después de la intervención dietética) en la presión arterial y en los parámetros de adiposidad. Después de la pMRHD, el peso corporal, el índice de masa corporal (IMC), la masa grasa, la circunferencia de la cintura y la presión arterial disminuyeron en ambos genotipos. El porcentaje de reducción de peso a los tres meses fue de 9,0 (5,3-9,7%) en portadores de alelos no G y una pérdida de peso similar en portadores de alelos G 8,7 (6-11%). La mejoría del peso corporal (-8,8 ± 2,3 kg vs. -8,6 ± 1,9 kg; p = 0,21), IMC (-3,3 ± 0,5 kg/m2 vs. -3,2 ± 0,6 kg/m2; p = 0,32), la masa grasa (-5,0 ± 1,1 kg vs. -4,9 ± 1,1 kg; p = 0,31) y la circunferencia de la cintura (-7,5 ± 1,2 cm vs. -9,0 ± 1,3 cm; p = 0,42) fueron similares en los portadores del alelo G que en los no portadores. La mejora en la presión arterial fue similar en ambos genotipos (tabla 2).
Parámetros antropométricos y presión arterial (media ± DE)
| Parámetros | n = 44 | pTiempo | pGenotipo x tiempoGenotipo PreGenotipo Post | ||||
|---|---|---|---|---|---|---|---|
| AA (n = 27) | AG + GG (n = 17) | ||||||
| Basal | 3 meses | ptiempo | Basal | 3 meses | |||
| IMC | 38,6 ± 2,1 | 35,3 ± 4,0 * | p = 0,005 | 38,1 ± 2,0 | 34,9 ± 1,1* | p = 0,004 | p = 0,33p = 0,32p = 0,39 |
| Peso (kg) | 97,7 ± 10,5 | 88,9 ± 9,0$ | p = 0,004 | 97,9 ± 9,1 | 89,3 ± 8,1$ | p = 0,002 | p = 0,28p = 0,36p = 0,42 |
| Masa grasa (kg) | 43,7 ± 5,1 | 38,7 ± 6,0# | p = 0,007 | 41,9 ± 5,0 | 37,0 ± 4,1# | p = 0,005 | p = 0,37p = 0,53p = 0,68 |
| CC (cm) | 116,1 ± 7,1 | 108,6 ± 7,0& | p = 0,02 | 115,8 ± 7,0 | 106,8 ± 5,1& | p = 0,01 | p = 0,45p = 0,43p = 0,56 |
| TAS (mmHg) | 134,3 ± 7,1 | 118,1 ± 5,0** | p = 0,01 | 136,9 ± 6.0 | 120,5 ± 5,2** | p = 0,02 | p = 0,47p = 0,34p = 0,45 |
| TAD (mmHg) | 80,5 ± 6,0 | 76,5 ± 3,2 *** | p = 0,02 | 78,1 ± 4,1 | 75,4 ± 2,0*** | p = 0,03 | p = 0,53p = 0,60p = 0,71 |
Para analizar el efecto de la intervención en cada grupo p < 0,05 (
TAD) (p tiempo). Un valor para la interacción (p tiempo-grupo) que informa si el efecto de la intervención es el mismo para los dos grupos; t de Student para el análisis de las diferencias diferencias entre los dos grupos en la variable principal (antes y después) (p genotipo pre y post).
IMC: índice de masa corporal; TAD, tensión arterial diastólica; TAS, tensión arterial sistólica; CC, circunferencia cintura; DE: desviación estándar.
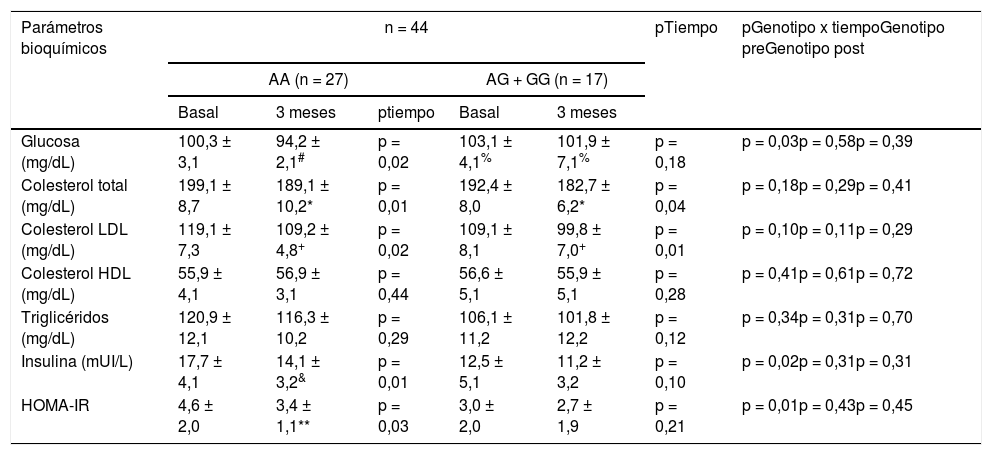
La tabla 3 muestra los cambios en los parámetros bioquímicos. Después de la intervención dietética con pMRHD, los sujetos sin alelo G mostraron una mejora significativa en los niveles de glucosa en ayunas (AA vs. AG + GG) (-6,1 ± 1,4 mg/dL vs. -1,2 ± 0,7 mg/dL; p = 0,01), ayuno niveles de insulina (-3,6 ± 0,2 mU/L vs. -1,3 ± 0,6 mU/L; p = 0,02) y HOMA-IR (-1,2 ± 0,2 unidades vs. -0,3 ± 0,2 unidades; p = 0,01). Los niveles de glucosa en plasma en ayunas fueron más altos en los portadores de alelos G que en los portadores de alelos no G (delta en el tiempo basal; -3,1 ± 0,2 mg/dL; p = 0,02) y (delta a los tres meses: -5,7 ± 0,4 mg/dL; p = 0,01). Finalmente, la mejora en los niveles de colesterol LDL y colesterol total fue similar en ambos genotipos (tabla 3).
Parámetros bioquímicos (media ± DE)
| Parámetros bioquímicos | n = 44 | pTiempo | pGenotipo x tiempoGenotipo preGenotipo post | ||||
|---|---|---|---|---|---|---|---|
| AA (n = 27) | AG + GG (n = 17) | ||||||
| Basal | 3 meses | ptiempo | Basal | 3 meses | |||
| Glucosa (mg/dL) | 100,3 ± 3,1 | 94,2 ± 2,1# | p = 0,02 | 103,1 ± 4,1% | 101,9 ± 7,1% | p = 0,18 | p = 0,03p = 0,58p = 0,39 |
| Colesterol total (mg/dL) | 199,1 ± 8,7 | 189,1 ± 10,2* | p = 0,01 | 192,4 ± 8,0 | 182,7 ± 6,2* | p = 0,04 | p = 0,18p = 0,29p = 0,41 |
| Colesterol LDL (mg/dL) | 119,1 ± 7,3 | 109,2 ± 4,8+ | p = 0,02 | 109,1 ± 8,1 | 99,8 ± 7,0+ | p = 0,01 | p = 0,10p = 0,11p = 0,29 |
| Colesterol HDL (mg/dL) | 55,9 ± 4,1 | 56,9 ± 3,1 | p = 0,44 | 56,6 ± 5,1 | 55,9 ± 5,1 | p = 0,28 | p = 0,41p = 0,61p = 0,72 |
| Triglicéridos (mg/dL) | 120,9 ± 12,1 | 116,3 ± 10,2 | p = 0,29 | 106,1 ± 11,2 | 101,8 ± 12,2 | p = 0,12 | p = 0,34p = 0,31p = 0,70 |
| Insulina (mUI/L) | 17,7 ± 4,1 | 14,1 ± 3,2& | p = 0,01 | 12,5 ± 5,1 | 11,2 ± 3,2 | p = 0,10 | p = 0,02p = 0,31p = 0,31 |
| HOMA-IR | 4,6 ± 2,0 | 3,4 ± 1,1** | p = 0,03 | 3,0 ± 2,0 | 2,7 ± 1,9 | p = 0,21 | p = 0,01p = 0,43p = 0,45 |
Diferencias estadísticas p < 0,05, en cada grupo de genotipo (# glucosa, & insulina,
HOMA-IR) (p tiempo). Un valor para la interacción (p tiempo-grupo) que informa si el efecto de la intervención es el mismo para los dos grupos; t de Student para el análisis de las diferencias entre los dos grupos en la variable principal (antes y después) (p genotipo pre y post).
HOMA-IR: homeostasis model assessment; DE: desviación estándar.
En este estudio mostramos cómo los sujetos con obesidad sin alelo G del SNP rs7756992 presentaron una mejoría en los niveles de glucosa basal en ayunas, niveles de insulina y HOMA-IR que los portadores del alelo G tras una dieta hipocalórica de reemplazo parcial de comida (pMRHD). Los portadores del alelo G mostraron niveles más altos de glucosa basal en ayunas que los no portadores del alelo.
En el mismo sentido de empeoramiento del metabolismo glucémico por este alelo de riesgo que hemos encontrado en nuestro trabajo, ya hay datos en la literatura. Un estudio anterior11 demostró que los portadores del alelo G de rs7756992 tienen una respuesta de insulina 22% menor que los no portadores del alelo G con carga de glucosa estándar. Chistiakov et al.19 mostraron niveles significativamente reducidos de insulina sérica a las 2 h en respuesta a la estimulación de glucosa en pacientes diabéticos y pacientes no diabéticos con alelo G de esta variante CDKAL1. Este alelo de riesgo está relacionado con una homeostasis de células beta deteriorada y células beta reducidas. El efecto nocivo de este alelo de riesgo sobre el metabolismo glucémico se ha demostrado no solo en adultos, sino también en niños20. Por otra parte, este efecto negativo puede comenzar ya en la edad fetal, debido a que se ha demostrado una asociación del alelo G con bajo peso al nacer, un factor de riesgo independiente conocido para la obesidad y la diabetes mellitus tipo 221. Dado que esta variante de CDKAL1 está relacionada con problemas en la secreción de insulina, un factor de crecimiento fetal bien conocido, la reducción de la secreción de insulina fetal en el útero produce este bajo peso al nacer22.
Para explicar nuestra peor respuesta en los niveles de glucosa en ayunas, niveles de insulina en ayunas y resistencia a la insulina después de la pérdida de peso en los portadores del alelo G, podemos proponer una hipótesis fisiopatológica. Podríamos plantear la hipótesis de que es probable que esta asociación sea secundaria a una sensibilidad reducida de las células beta y a una respuesta de insulina disminuida23. Una sensibilidad reducida a la glucosa en las células beta indica una disminución de la secreción de insulina, en este caso en sujetos con obesidad con resistencia periférica a la insulina. Además, esta disminución de la sensibilidad a la glucosa se relacionó con la progresión a una tolerancia anormal a la glucosa24, como hemos podido verificar en pacientes con obesidad que portan el alelo G de nuestro estudio, que tenían niveles de glucosa en ayunas más altos, antes y después de la pérdida de peso, secundario a la intervención nutricional.
Hasta donde sabemos, este es el primer estudio que evalúa en pacientes con obesidad el efecto de esta variante genética en los efectos glucémicos secundarios a la pérdida de peso después de una intervención dietética. En la literatura, algunos estudios previos han evaluado el efecto de los fármacos antidiabéticos y esta variante genética. Por ejemplo, Chistiakov et al.19 informaron una mejor glucemia posprandial después de sulfonilureas (SU) en pacientes con riesgo de alelo G de rs7756992. Del mismo modo, Schroner et al.25 informaron que la glucemia en ayunas después de la SU también se redujo más en sujetos con alelo G. Los mecanismos subyacentes a esta relación entre el alelo de riesgo de rs7756992 y la respuesta terapéutica a la SU no están claros. La mejora de los canales KATP y el ATP generado por las mitocondrias se ha implicado en estas acciones19,25, con una mejor respuesta en pacientes que portan el alelo G. El efecto negativo del alelo G en nuestro trabajo es solo en el control glucémico, ya que pudimos ver cómo mejoró el perfil lipídico en pacientes de ambos genotipos,
Las limitaciones de nuestro estudio son la inclusión en el ensayo de sujetos con obesidad sin diabetes mellitus tipo 2 establecida. En segundo lugar, solo analizamos un SNP del gen CDKAL1, por lo que otras variantes genéticas en este gen u otros podrían estar relacionadas con los resultados. Tercero, la ausencia de un grupo control sin dieta podría ser un sesgo. Cuarto, no hemos determinado la respuesta de glucosa en sangre posprandial. Finalmente, la ingesta dietética autoinformada no es del todo fiable y podría incluir un sesgo de informes insuficientes o excesivos. La fuerza de nuestro estudio estuvo en su diseño como un ensayo intervencionista con alta adherencia a un reemplazo parcial de comida y las frecuencias del alelo G estaban de acuerdo con los estudios previos26,27.
En resumen, nuestros datos sugieren que la variante genética (rs7756992) del gen CDKAL1 está asociada con el estado glucémico después de una intervención de reemplazo parcial de comida, con una mayor mejoría de la glucosa en ayunas, la insulina y el HOMA-IR en sujetos sin alelo G. Los mecanismos subyacentes exactos de esta variante genética del gen CDKAL1 que modifican los niveles de glucosa y la sensibilidad a la insulina aún no están claros y deben investigarse en estudios adicionales para evaluar el papel en la diabetes mellitus28 y sus complicaciones29.
Declaración de éticaEste estudio intervencionista aprobado por nuestro Comité de Ética (Comité HCUVA 07/2018). Los pacientes fueron informados de su derecho a retirarse del estudio sin comprometer su tratamiento y todo el consentimiento firmado e informado.
Contribuciones de autorDaniel de Luis escribió el artículo, diseñó el estudio. Olatz Izaola escribió el artículo y realizó una evaluación nutricional. David Primo realizó evaluaciones bioquímicas. J.J. López realizó evaluaciones nutricionales y trabajos estadístiscos.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
