


Henri Jules Debray (1827-1888), a student of Henri Sainte-Claire Deville, carried on a large number of important researches in the areas of mineralogy, crystallization, synthetic reproduction of natural crystals, preparation of beryllium and its salts, and particularly, in the metallurgy, preparation of the pure metals, and synthesis of derivatives of the metals of the platinum group.
Henri Jules Debray (1827-1888), un alumno de Henri Sainte-Claire Deville, realizó un gran número de investigaciones importantes en las áreas de mineralogía, cristalización, reproducción sintética de cristales naturales, preparación del berilio y sus sales, y particularmente, en la metalurgia, preparación de los metales puros y síntesis de derivados de los metales del grupo del platino.
Very little information is available about the early life of Henri Jules Debray (1827-1888). A detailed description of his scientific activities was written by one of his former students, Alfred Ditte (1843-1908) (Ditte, 1889). Henri Jules Debray was born in Amiens, France, on 26 July 1827; in 1847, after finishing his basic education, he entered the École Normale Supérieure in Paris, where he studied and worked as préparateur under Henri Sainte-Claire Deville (1818-1881), and also pursued his doctoral studies. In 1850 he began teaching at the Lycée Charlemagne, and in 1855 he received his doctorate (docteur ès Sciences) from the Faculté des Sciences, after successfully defending his thesis about beryllium (glucinium) and its compounds (Debray, 1855). The salts of beryllium had not been studied because of the scarcity of the metal and the difficult process to separate it. Debray developed a method for preparing the metal in amounts large enough to be able to make a detailed study of its compounds, and prepared beryllium as globules and thin laminæ. His results indicated that beryllium and aluminum had very similar properties (Ditte, 1889). His first post-doctoral papers, done in collaboration with Deville, were about the platinum metals and their treatment by the dry procedure (Deville and Debray, 1857, 1859b). From 1855 to 1868 Debray served as assistant at the École Normale Supérieure, and from 1868 on he began teaching at the École Polytechnique, a position he led until his death on 19 July 1888. After Deville illness, Debray replaced him at the Chair of General Chemistry of the Faculté des Sciences, and In 1888 Ditte (1843-1908) replaced Debray at this position. On February 16 1877 Debray was elected to the Académie des Sciences (chemistry section).
Among other positions held, Debray served as assayer at the Mint, member of the Bureau de Consultation des Arts et Métiers, and Vice President of the International Committee of Weights ad Measures.
Debray worked extensively on many different subjects. He showed that the dissociation of a solid, with production of a gas phase, followed the same laws as the vaporization of a liquid; he also showed that the usual laws of dissociation were not applicable as such to the case of hydrates. Debray perfected Deville’s method for synthesizing many natural oxides (Deville, 1861), and prepared a large number of them, for example, azurite, atacamite, arseniates, tungstic acid and tungstates, phosphates, etc. He also showed that the crystalline form of this oxides was mainly determined by the temperature of at which they were synthesized. His most important works were related to platinum and the metals that accompanied it.
Scientific contributionDebray published over 60 papers, several of them in the form of booklets and books (Debray, 1861, 1863ab, 1871, 1873b; Deville and Debray, 1859a). As customary of all candidates to a position in the Académie des Sciences, he also published a brochure describing his scientific work and results (Debray, 1876). He is best known for his extensive researches in the area of metallurgy, crystallization, dissociation, preparation of the pure elements and their derivatives, of metals of the platinum group. Some of his work with Deville about platinum and the platinum metals group has been presented in another publication and will not be repeated here (Wisniak, 2004).
BerylliumDebray’s first major publication was a result of his doctoral thesis about beryllium and its compounds (Debray, 1855). Scientists, such as Martin-Heinrich Klaproth (1743-1817) (Klaproth, 1797) and Johann Jacob Bindheim (1750-1825), had analyzed emerald and aquamarine (beryl) and established that they had a very similar chemical composition, while René-Just Haüy (1743-1822) had established a complete correspondence between the structure, hardness, and density of the two minerals and concluded that they were the same material. To verify these facts, Haüy requested from Louis Nicolas Vauquelin (1763-1829) to analyze both minerals and see if they had the same composition. Vauquelin first fused beryl with potassium hydroxide, separated different fractions, treated them with aqueous HCl, potassium carbonate, nitric acid, etc., and eventually was left with a residue that he realized was the carbonate of the oxide of a new earth, which he called terre de béril (Vauquelin, 1798). According to Vauquelin, emerald contained, by weight, 64% silica, 29% alumina, 2% calcium carbonate, and about 2% water, while beryl contained 69% silica, 21% alumina, 8% of the new material, and about 05% iron. Additional analytical work led him to find that the new material was also present in emerald and that the difference in color between the two stones was simply due to the coloring impurities present in each. Vauquelin, noting that many of the compounds derived from the new earth had a sweet taste, wrote that he did not believe that he had to give a definite name to this before he knew its properties better. Besides, he would be happy to have the advise of his colleagues. The editors of the journal picked up this suggestion and proposed naming it glucine (from the Greek: to sweeten). The corresponding name of the element would have then be glucyum, but Martin-Hein-rich Klaproth (1743-1817), Wöhler, and others, preferred the names beryllia and beryllium due to fact that yttria also formed sweet salts. Later, this earth was found in many minerals, among them, cymophane (chrysoberyl), phenacite (a beryllium silicate), gadolinite (a dark green or greenish-black silicate mineral, Be2FeY2Si2O10), leucophanite [a soro-silicate mineral, (Na, Ca)2BeSi2(O·OH·F)7], and helvine (a mineral consisting of silica, beryllium, manganese, iron, and sulfur).
Friedrich Wöhler (1800-1882) and Antoine Bussy (1794-1882) independently isolated beryllium in 1828 by reacting beryllium chloride with potassium. The element was described as a deep grey powder that looked like a metal precipitated as very small particles; burnished, it assumed a dark metallic brilliancy. The strong heat required for its preparation did not cause particle agglomeration, suggesting that it would difficult to melt. Beryllium did not oxidize in air at room temperature but burned brilliantly when heated red; the combustion was even more active when done in the presence of pure oxygen. The formation of beryllium sulfide was accompanied by incandescence (Bussy, 1828; Wöhler, 1828).
In the first part of his work, Debray gave a short description of the history of the mineral and the element, prepared pure beryllium using Wöhler’s method, and repeated many of the latter experiments, with conflicting results. According to Debray, beryllium was a white metal of density 2.1. It was the lightest of all the metals that did not decompose water when heated above 100°C. Beryllium could be melted at the oxidation flame of the blowtorch but did not catch fire in an atmosphere of pure oxygen; in both cases the metal became covered with a light layer of oxide, which prevented further oxidation. Debray believed its melting point was below that of silver (960.5°C). Adding NaCl as a flux could easily agglomerate the metal globules. He found that the beryllium he had prepared remained unchanged in an atmosphere of sulfur vapors, showing that no sulfide was formed. Nevertheless, there were indirect methods, which could be used to prepare easily this sulfide (see below). Reaction of beryllium with chlorine, iodine, silicon, water, inorganic acids, ammonia, etc., also produced results different from those reported by Wöhler, which led Debray to believe that the beryllium prepared by Wöhler was in an impure state and that it probably contained residues of potassium and platinum (Debray, 1855).
Glucine (beryllium oxide) was normally prepared from Limoges’s emerald using Berzelius’s elaborate method, which was limited to very small amounts of material. Debray developed a much simple procedure, which allowed preparation of glucine in much larger quantities. His process was based in melting the emerald with quicklime, treating the product with nitric acid, evaporating the liquid to eliminate the excess of acid, and then decomposing the nitrates of beryllium aluminum, and iron by calcination. The residue was composed of insoluble silica, alumina, glucine, and iron sesquioxide. The glucine was separated by successively washing this residue with several reagents, such as an ammonium salt, boiling nitric acid, ammonium carbonate, etc. The resulting beryllium carbonate was converted into beryllium oxide by calcination, or used as such to prepare many beryllium derivatives (beryllium chloride, iodide, fluoride, sulfate, carbonate, and oxalate, double carbonate of beryllium and potassium, double carbonate of beryllium and ammonium, double sulfate of beryllium and ammonium, beryllium and potassium oxalate, and beryllium and ammonium oxalate). Debray gave a detailed account of the preparation and properties of each of the salts, including beryllium oxide (Debray, 1855).
Copper mineralsHenri Hureau de Sénarmont (1808–1862) had tried vainly to reproduce azurite (the blue copper carbonate), operating at about 200°C by a method that had allowed him to obtain the carbonates of magnesium, iron, manganese and zinc (de Sénarmont, 1849). Debray was able to carry on the synthesis of the salt by reacting at room temperature, in a closed tube, a solution of cupric nitrate with an excess of calcium carbonate. He observed that the calcium carbonate became covered with a layer of a green material while the solution lost its color. The layer eventually transformed into rounded protuberances of blue copper carbonate (azurite). According to Debray, the reaction took place in two stages. In the first one the calcium carbonate slowly transformed into the tribasic copper nitrate and CO2
and once the neutral copper nitrate had disappeared, the gas formed calcium bicarbonate, which reacted slowly with the tribasic nitrate of copper and transformed it into azurite: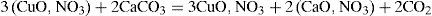
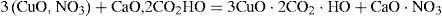
Substitution of calcium carbonate by alkaline carbonates resulted in the formation of a double carbonate of copper and sodium (or potassium). Potassium bicarbonate also yielded a well- crystallized product, which was easily decomposed by water. Debray reported that no reaction took place when contacting ordinary copper carbonate or malachite (copper carbonate hydroxide) with CO2 at high pressures (10 to 14 atm) (Debray, 1859).
Debray also succeeded in reproducing crystals of an hydrated oxychloride of copper having the composition of atacamite, by two different methods: (a) heating to 200°C a mixture of tribasic copper nitrate with a saturated solution of NaCl. According to Debray, no reaction took place when the copper nitrate was replaced by other copper salts such as the basic sulfate [brochantite, CuSO4·3Cu(OH)2] and the basic phosphate [libethenite, Cu2(PO4)(OH)]; and (b) by reacting in a water bath ammonia copper sulfate with an excess of concentrated aqueous NaCl. The ammonia was driven out and the resulting crystalline precipitate was easily washed with water to eliminate the remaining NaCl. The resulting product contained 58.4% copper and 16.3% chlorine, compared with 59.5% copper and 16.5% chlorine for atacamite. Debray found remarkable the fact that the reaction occurred only in the presence of NaCl or KCl (Debray, 1867).
Tungsten derivativesIn 1862 Debray reported an easy method for obtaining crystalline anhydrous tungstic acid by passing a stream of HCl over a mixture of sodium tungstate and sodium carbonate heated red in a porcelain tube. In the following reaction, the tungstic acid was released by the HCl and crystallized over the NaCl formed, as dark green crystalline rectangular prisms. Carrying on the reaction at white red resulted in the crystals being deposited on the walls of the tube as octahedral crystals (Debray, 1862). The apparent volatilization of tungstic acid in HCl was another example of the phenomenon discovered by Deville regarding the action of this acid on amorphous oxides: it transformed them into crystalline substances (Deville, 1861). This required the acid reacting with the oxide to produce a chloride and water, which then reacted between them to regenerate the acid and produce a crystalline oxide. If the stream of acid was slow, there was no transport of the products and the forward and inverse reaction occurred on the same place. If the flow of HCl was fast, the water vapor and chloride formed were carried on, and the inverse reaction occurred further on (Debray, 1862).
The same reaction carried on with calcium tungstate transformed the salt into the neutral calcium tungstate (scheelite, CaWO4), which crystallized in the excess of calcium chloride as regular octahedrons, the same as the natural product. Iron tungstate was produced in the same manner (Debray, 1862).
A following publication described the synthesis of the different chlorides of tungsten and some of their properties (Debray, 1865). Passing a stream of dry chlorine over tungsten heated dark red, produced deep red vapors that condensed as a dark gray solid composed of tungsten perchlo-ride (WCl3) and tungsten sub-chloride (W2Cl5). Distillation of the mixture in a stream of chlorine produced the perchlo-ride almost pure. Distillation of a mixture of tungsten perchloride and dry oxalic yielded a blend of two oxychlorides, a red one having the formula WOCl2, and a slightly yellow one corresponding to WO2Cl. The first oxychloride could also be obtained by heating a mixture of anhydrous tungstinic acid with tungsten perchloride
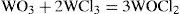
Debray also reported what he thought were the approximate densities of the vapors of these chlorides: 11.50 for WCl3 and 10.74 for WOCl2 at the temperature of boiling mercury vapor (359.6°C), and 11.69 to 11.89 and 10.27 respectively, at the boiling temperature of sulfur (444.6°C) (Debray, 1865).
Molybdenum and derivativesAs told by Debray, the sodium acid molybdate hydrate sold by the local commerce under the name of German molybdic acid, could be used easily for the preparation of molybdic acid and various of its compounds (Debray, 1858). His procedure was as follows: A mixture of acid molybdate of soda and ammonium chloride was heated to redness in an earthen crucible. The resulting mixture contained NaCl, molybdic acid, metallic molybdenum, and molybdenum sulfide. The latter originated from the decomposition of sodium sulfate contained in German molybdic acid. The soluble salts were easily eliminated from the mixture by water washes; at the beginning, the water, strongly charged with salts, passed over colorless, and afterwards it, it assumed a bluish tint which showed the solution of a very small quantity of oxide.
On account of their state of extreme division, the molybdenum and its oxide and sulfide thus obtained, burned with extreme facility, even below red heat. This fact allowed an easy transformation into acid; it was enough to roast the mixture in a cupel at a very low temperature, in order not to lose any of the volatile acid (volatile at a red heat). The product was then put into a platinum boat, heated to a bright red, and under the influence of the feeble thermal current of air produced in the tube, the molybdic acid volatilized, and deposited on the upper part of the tube in beautiful crystalline plates (Debray, 1858).
Metallic molybdenum was obtained by reducing the acid with hydrogen first at a low temperature, and then at white heat to complete the operation. The metal thus obtained was in a state of extreme division; it did not present any trace of fusion, or even of agglomeration, as platinum did in the same circumstances. Debray tried unsuccessfully to fuse the molybdenum by several means, including those used by Deville to fuse platinum. Eventually he succeeded by heating the molybdenum particles, mixed with a flux such as calcium aluminate, in a charcoal crucible, protected by a lime crucible, by means of the oxy-hydrogen blowpipe. This allowed him to raise the temperature to the fusing point of rhodium (1995°C). The fused metal obtained was not absolutely pure; it contained from 4 to 5% of charcoal, originating from the crucible, which must have increased its fusibility. It was white, with a luster approaching that of silver, it easily scratched glass and topaz but was not affected by the hardest steel and could not be polished with boron powder. Its density was 8.6, that is, half that of tungsten, and its chemical properties did not differ from that of the divided metal.
Debray then described the preparation of a chlorine derivative. Passing a stream of HCl over molybdic acid heated to 150° to 200°C, generated a white crystallized compound, very volatile, very soluble in water, and decomposed by heat into HCl and molybdic acid. Chemical analysis indicated that is composition corresponded to the formula MoO3·HCl. Phosphoric acid was found to dissolve a considerable quantity of non-volatilized molybdic acid. Evaporation of the resulting solution did not lead to crystallization, only to a syrupy liquid. Saturating the solution with a solution of ammonia, followed by cooling, led to the formation of beautiful crystals of a double salt (Debray, 1858).
Another paper described in detail the combinations of molybdic and phosphoric acids (Debray, 1868b). It was known that mixing a nitric solution of ammonium molyb-date with phosphoric acid produced a yellow material almost insoluble in all the acids. The precipitate contained about 89% molybdic acid, 4% phosphoric acid, and some water and ammonia. Boiling it with aqua regia eliminated the ammonia and evaporation of the remaining liquor precipitated beautiful yellow double oblique prisms, composed of one equivalent of anhydrous phosphoric acid (P2O5), 20 equivalents of anhydrous molybdic acid (MoO3), and 13.3% of water. These crystals, very soluble in water, could form two additional hydrates, containing 19.6% and 23.4% water, respectively. The small amounts of phosphoric acid combined with molybdic acid in these hydrates (3.7 to 4.1%), was enough to produce a substantial change in their physical and chemical properties; for example, molybdic acid could easily form an hydrate which produced colorless non crystallizable solutions; while the hydrates of phosphomolybdic acid were yellow and easily crystallizable (Debray, 1868b).
Debray described the preparation and properties of a large number of phosphomolybdates and double salts with nitric acid. For example, the composition of the yellow phosphomolybdates of potassium, thallium, and ammonia, could be represented by the general formula (3RO, PO5, 20MoO3). All were well-defined compounds, not mixtures, because they could be easily crystallized. Treating a solution of silver nitrate with phosphomolybdic acid precipitated microscopic crystals having the composition (7AgO, PO5, 20MoO3+24H2O). This salt dissolved in dilute nitric acid; evaporation of the resulting solution yielded microscopic yellow crystals having the composition (2AgO, PO5, 20MoO3+7H2O) (Debray, 1868b).
According to Debray, the existence of phosphomolybdic compounds and the frequent association of molybdic and vanadic acids in natural compounds, led attributing to molybdic acid the formula Mo’O5 (where Mo’=(5/3)Mo). The constitution of phosphomolybdic acids was quite appropriate for this substitution because the formulas (PO5, 20MoO3) and (PO5, 5MoO3) became (PO5, 12Mo’O5) and (PO5, 3Mo’O5), respectively. Nevertheless, the value of the density of the vapors of molybdenum chloride (easily measurable) contradicted this formulation (Debray, 1868c). Debray prepared molybdenum chloride by the direct action of chlorine on the slightly heated metal; the product, distilled in a current of dry CO2 (to eliminate the excess of chlorine), had a deep green color; it melted at 194°C, and boiled at 268°C releasing deep red vapors. Analysis of the chloride indicated that it contained 35 to 35.2% of molybdenum, a composition appropriate to the formula Mo2Cl5. Measurement of the density of the vapors at 350°C gave values between 9.40 and 9.53, which compared very well with the theoretical of 9.47 for the formula Mo2Cl5 (against 15.8 for the formula Mo’O5).
Several scientists had reported the values 46 or 48 for the equivalent of molybdenum. Debray prepared pure molybdic acid by subliming it in a platinum tube. Since the sublimed acid was extremely bulky, Debray dissolved it in ammonia and then calcined the resulting salt. By this means he obtained an acid both dense and free from lower oxides. The acid was then reduced to molybdenum by heating in a current of hydrogen.
The equivalent deduced from the amount of metal obtained from a given weight of molybdic acid was, in three experiments, 48.03, 48.04, and 47.84, respectively. Preparing silver molybdate and determining the amount of silver present in it confirmed these results (The value 48 corresponds to an atomic mass of 96, which is very close to the accepted value 95.84) (Debray, 1868c).
Previously Debray had shown that phosphoric acid combined with molybdic acid in the ratio of 1:20 equivalents and that the resulting yellow phosphomolybdic acid was soluble in water and precipitated potassium and ammonia from their acid solutions (Debray, 1868b). In an additional publication, he showed that a similar phenomenon occurred between arsenic acid and molybdic acid: an arseniomolybdic acid was formed containing the two acids in the ratio of 1:20 equivalents. The white acid crystallized as right rhomboidal prisms; it was stable in the presence of acids and combined with bases without decomposition. Neutralization with an alkali produced a white gelatinous precipitate, little soluble in cold water, and soluble in excess acid or alkali. Debray reported the preparation and properties of sodium and ammonium arseniomolybdate (Debray, 1874a).
Osmium and iridiumIn a memory published in 1869, Deville and Debray described a procedure for the reproduction of ferriferous platinum and ruthenium sulfide (RuS2, laurite) (Deville and Debray, 1879). Their method consisted in melting in an earthen pot a mixture of finely divided platinum (or ruthenium) with iron pyrite and borax. If the temperature was not too high the product contained the sulfides of platinum or of ruthenium, crystallized within the mass of iron sulfide. Treatment with aqueous HCl eliminated the iron and each sulfide was then purified using the appropriate reagents. The artificial laurite crystallized as regular octahedrons or as cubic crystals, the same as natural laurite. Treatment with concentrated HCl, cold at first, and boiling towards the end of the operation, dissolved all the iron sulfide and left a solid formed by the two sulfides of ruthenium, a black one soluble in dilute nitric acid, and a bluish one, insoluble in all acids, including aqua regia. The nitric acid solution was first treated with concentrated KOH to eliminate the gelatinous silica coming from the glass, and then with HF to eliminate the small fragments of the earthen pot. Heating the purified material to a high temperature decomposed the sulfur and left metallic ruthenium crystallized as small cubic crystals (Deville and Debray, 1879).
Platinum sulfur was prepared and purified by the same method and shown to have the composition PtS. It crystallized as gray needles insoluble in aqua regia. Heating this sulfur to a very high temperature left a crystalline matter of ferriferous platinum, containing about 11% weight iron, soluble in aqua regia, and having no magnetic properties. Its composition was the same as some natural platinum alloys (Deville and Debray, 1879).
In a continuation of this paper, Debray reported the behavior of iridium and osmium under the same circumstances (Debray, 1882). Heating a mixture of iridium with iron pyrite produced a slag that treated with diluted HCl left a deposit of crystallized iridium mixed with a light black sulfide. This sulfide was easily dissolved with nitric acid, leaving the iridium in a medium from which it crystallized and transformed into a sulfide. The iridium sulfide decomposed at high temperature and left iridium as regular hexagonal crystals, mixed with 1 to 2% iron. Osmium behaved in the same manner. Melting the metal at high temperature with pyrite and a little of borax gave a slag, which treated with HCl left a mixture of crystalline osmium and a black sulfurized substance. Again, treating the slag with nitric acid eliminated the latter and left the osmium crystals alone, without traces of iron (Debray, 1882).
Osmium and iridium were known to be present (as osmides) in variable proportions in natural platinum minerals. For this reason Debray decided to study the action of iron py-rite on such mixtures. He melted a large amount of pyrite with one part of amorphous osmium and 1, 2, or 3 parts of amorphous iridium, and treated the resulting slag successively with HCl and HNO3. In every case he obtained a homogenous crystalline residue composed of hexagonal laminæ, similar to those of the natural osmides. These crystals did not have the original proportion of osmium to iridium because of the accompanying sulfides, which were formed simultaneously. The relative proportion of the two metals was, mostly, a function of the operating temperature.
The results indicated clearly that osmium and iridium could crystallize together in any proportion, without modifying the form of their combination; hence, they were isomorphs belonging to the cubic system (Debray, 1882).
The behavior of rhodium was substantially different. Melting finely divided rhodium with 20 to 30 times its weight of iron pyrite produced a slag that treated with HCl left a deposit of blackish flakes, seemingly semi-metallic. This deposit was completely soluble in diluted nitric acid, giving a strongly colored solution. According to Debray, this product was not a sulfide because dried at 100°C in dry air or under vacuum, retained 9.6% of water and 8.6% of oxygen as integral parts of it. Upon heating, the water and the oxygen were liberated with the oxygen becoming SO2. The residue was a rhodium protosulfide, RhS, containing traces of iron (0.7%), which was not attacked by aqua regia. Debray believed that the reaction of rhodium with iron pyrite formed a rhodium sesquisulfide that afterwards, at 100°C decomposed according to (Debray, 1883)
Dissociation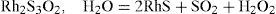
In 1864 Deville demonstrated experimentally that certain gaseous or volatile compounds decomposed partially and progressively as the temperature increased. The resulting product was a mixture formed by the gases originating from the starting substance and the part of it that had decomposed. At a given temperature, the pressure of this mixture was constant but increased as the temperature was augmented. Deville had named the phenomenon dissociation (Deville, 1864-1865). In a paper published in 1868, Debray was intent in proving that dissociation also took place in solid bodies formed by the combination of two substances, one fixed and the other volatile, and that the phenomenon continued to be controlled by the temperature (Debray, 1868a).
To illustrate his hypothesis, Debray studied the thermal decomposition of Iceland spar (a crystalline variety of calcium carbonate) in a tube made of glass or varnished porcelain, and heated successively with vapors of mercury (350°C), sulfur (440°C), cadmium (860°C), and zinc (1040°C), connected to a vacuum system (for extracting the gases produced) and a pressure measuring device. The spar decomposition started at above 350°C, it was still very small at 440°C, and become quite appreciable at 860°C when the CO2 re-leased exerted a pressure of 85mmHg. If the pressure was lowered below this value (for example, by absorbing the CO2 in KOH), the decomposition of the carbonate was reinitiated, and CO2 was released until the pressure reached again 85mmHg. At 1040°C the decomposition of the spar was quite considerable and stopped when the pressure of the CO2 reached 520mmHg. The pressure developed by the gas at a certain temperature was independent of the state of decomposition of the carbonate because if the latter was replaced by quicklime and the apparatus filled with CO2 at a pressure lower than that required to saturate it completely, the pressure dropped to 85mmHg if the temperature was 860°C and to 520mmHg if it was 1040°C. Hence, this value represented the equilibrium pressure of the system at the indicated temperature and was independent of the state of decomposition of the carbonate (Debray, 1868a).
Debray pointed out the strong analogy that existed between the dissociation phenomenon and that of vaporization of a liquid. In both cases achievement of a constant vapor (gas) pressure represented the necessary and sufficient condition for equilibrium. It was to be understood that if it was possible to evaporate completely a liquid at a given temperature, then it was equally possible to completely destroy a carbonate as long as the CO2 released was withdrawn at the rate it was formed (Debray, 1868a).
In a continuation of the above paper, Debray extended his findings to the efflorescence of hydrated salts, a dissociation phenomenon that had been studied by Deville (Deville, 1864-1865). Measurement of the pressure of the water vapor given off by a hydrated salt under vacuum indicated that it depended only on the temperature. If the hot hydrate was left to cool, the vapor pressure decreased because the salt absorbed back part of the water it had released; the new pressure was exactly the same attained if the cold hydrate was heated to the temperature in question. The phenomena of efflorescence or hydration were thus easily explained; the pressure of the air had no influence on the partial pressure of the water vapor formed. A salt would effloresce if the pressure of its vapors were higher than that of the humidity present in the air, and vice versa. Non-efflorescent salts owed this property to the fact that at ordinary temperatures the pressure of the water vapor they were able to release was always lower of that of the humidity commonly present in air. Heating sodium sulfate decahydrate to its fusion temperature (33°C) showed that the pressure of the water vapor released along the fusion process did not change. The same phenomenon was observed during the fusion of sodium carbonate decahydrate (34.5°C) (Debray, 1868a).
Debray recognized that the efflorescence of salts hydrates differed from the dissociation of calcium carbonate by a special feature. In the latter case the decomposition ended when the partial pressure of CO2 reached a given value at the temperature of the process. This was not the situation for a hydrate: the pressure of the water vapor released was not absolutely independent of the proportion of the water remaining in the salt. For example, sodium phosphate hydrated with 24 molecules of water (2NaO, OH, PO3+24H2O), lost most of its hydration water by exposition to air and the pressure of the salt was independent of its efflorescence state; thus a salt containing all of its hydration water (62.8%), and an effloresced salt containing nor more that 53 to 54% water, generated exactly the same vapor pressure, If the water content went below 50%, which corresponded to the hydrate (2NaO, OH, PO3+14H2O), a state that could be obtained by carrying on the crystallization below 31°C, the value of the vapor was sensibly lower. Hence, in the first stage of its decomposition, common sodium phosphate behaved like a combination of water and phosphate with 24 equivalents of water of hydration. This combination dissociated in the same manner as calcium carbonate, exerting a constant vapor pressure a given temperature. At the end of this stage, the salt with 14 equivalents of water also dissociated, but now with a smaller vapor pressure. Thus, the difference between the decomposition of a hydrated salt and calcium carbonate was that the latter did not present intermediate combination states between calcium oxide and calcium carbonate, as it existed between a anhydrous salt and its several hydrates (Debray, 1868a).
In 1874 Gustav Heinrich Wiedemann (1826-1899) published a memoir about the dissociation of the hydrates of the magnesium group (Wiedemann, 1874), where he claimed priority to the hypotheses that Debray had stated in the paper described above (Debray, 1868a). Wiedemann’s arguments were rejected by Debray because the papers referred to different hydrates and different operating procedures (Debray, 1874b).
Debray used the laws of dissociation and of pressure to explain some unexplained phenomena and to give a more plausible explanation for others. In 1873 Jacob Myers (Mijers) (Myers, 1873) reported his study about the thermal dissociation of red mercuric oxide (HgO). He found that between 150° and 293°C the pressure of the gas released was 2mmHg and increased to 8, 16, and 343mmHg, as the temperature went up 350°, 400°, and 500°C, respectively. The pressure did not seem to have an upper limit; it grew slowly as time went on. The decomposition occurred in a continuous manner becoming total after a sufficiently long period of time. Debray recognized the accuracy of the experiments carried on by Myers but rejected his conclusions. In order to study correctly the dissociation of any body it was necessary to heat it in a space having the same temperature at each of its points. This was not the situation in Myer’s equipment: the mercury vapors that separated condensed in a natural and continuous manner in the cold parts of the equipment and hence became isolated from any future further action of the oxygen. This fact disrupted the equilibrium existing between the decomposition pressure of the oxide and that of combination of the separate elements. More oxide decomposition had to take place to replace the mercury isolated from the action of oxygen, causing an increase of the partial pressure of the latter. Thus there was no a priori reason for the oxygen pressure not to increase indefinitely (Debray, 1873a).
In 1807 Humphry Davy (1778-1819) reported to the Royal Society the isolation of potassium by decomposing a fragment of slightly moistened potassium hydroxide by means of a voltaic cell (Davy, 1808). Davy’s procedure was expensive and allowed the preparation of only very small amounts of the metal, a fact that complicated the study of its properties and reactions. To obviate this problem Joseph-Louis Gay-Lussac (1778-1850) and Louis-Jacques Thenard (1777-1857) developed a new process based on the reaction between potassium hydroxide and metallic iron (Gay-Lussac and Thenard, 1808). Gay-Lussac and Thenard’s experiment presented a feature that the authors did not explain. In their process, iron turnings were heated to whiteness in a curved gun barrel and then melted potassium hydroxide was passed slowly over the ignited iron; the hydroxide decomposed into a mixture of potassium vapors, hydrogen, and oxygen. This reaction occurred only if the iron barrel was heated to a sufficiently high temperature. The potassium and the hydrogen left the apparatus while the oxygen became attached to part of the iron of the apparatus. Nevertheless, the iron in the hottest part was always found to be bright and unaltered because the oxygen reacted with the iron on the coldest part of the equipment. According to Debray, although KOH dissociated into its elements in the hottest section of the tube, at the place were the temperature was lower, the elements were in a thermal state that allowed their recombination (Debray, 1879). The iron absorbed the oxygen and the resulting oxide resisted reduction by the potassium thanks to the fact that the potassium (produced by the forward reaction) covered the metal with an impervious varnish. In 1870 Deville had proved that immersing in a dense hydrogen atmosphere a mass of iron incompletely oxidized, with its diverse parts at different temperatures, the oxygen would remain in the colder parts of the metal and abandon those that were hotter (Deville, 1870).
Deville and Debray studied the oxidation of the elements of the platinum group and found that the oxides of rhodium, palladium, and iridium, decomposed when heated to a temperature high enough and that that the reaction followed the usual dissociation laws. They considered in detail the dissociation of iridium oxide and measured the pressure of the released oxygen at temperatures between 823° and 1139°C; their results indicated that above 1000°C this pressure was about 203mmHg, while that of oxygen in the atmosphere was only about 152mmHg. This meant that above 1000°C iridium oxide decomposed in atmospheric air so that at this and a higher temperature, iridium was absolutely not oxidized in air (Deville and Debray, 1857).
It was known that copper heated in air transformed into the black oxide (CuO), without going through a lower oxidation state (an oxydule). This reaction took place at all temperatures between that of start of oxidation (< 350°C) and the temperature at which the pressure of dissociation of cupric oxide approximated that of oxygen in atmospheric air. Below this temperature, the black oxide decomposed and stopped when the partial pressure achieved the value of the external oxygen pressure. The resulting product was a molten mixture of the oxydule and the black oxide, in a proportion that depended on the temperature only. Cooling the mixture resulted in reoxidation, if it was sufficiently porous. Otherwise, the absorption of oxygen occurred only at a thin surface layer and the solid mixture maintained in its interior the average composition it had when in the molten state (Debray and Joannis, 1884). Debray and Joanis considered the particular case were the copper was in great excess with respect the oxygen, for example, during the preparation of nitrogen from air free of CO2 and water. In this process it was assumed that passing a stream of air through a column filled with copper turnings heated dark red, resulted in the total absorption of the oxygen. According to Debray and Joanis, when the experiment was carried below red heat, the oxide formed at the surface of the metal was the black one. This color allowed following the course of the reaction. But if a strong heating were carried in a glass tube, so that attain a temperature that dissociated the black oxide, the copper would blacken only at the entrance of the tube, a relatively cold location. It would go into the oxydule state in all places were the temperature was high enough to result in a sensible decomposition of the black oxide. A mixture of black oxide and metallic copper could only exist at a temperature were the black oxide begun to dissociate because the released oxygen combined with the copper to form the oxydule. The metallic copper would act like an aspiration pump, which would prevent the disengaged oxygen of reaching the dissociation pressure to limit the decomposition of the oxide; thus the copper would be completely converted into its lower oxidation state (Debray and Joanis, 1884).
Debray and Joanis also analyzed he intermediate situation were the oxygen present was not enough to completely oxidize the copper to cupric oxide but enough to provide a mixture of cuprous and cupric oxides. If the mixture was not totally molten, it was impossible to achieve an intimate mixture of both oxides. On cooling slowly, the red oxide and the black oxide were clearly separated. The red oxide was able to absorb the oxygen more completely than metallic copper (Debray and Joanis, 1884).
As a result of their work, Debray and Joanis developed a method allowing making a more complete vacuum in a tube without using the delicate and complicated methods then in use.
Another interesting case of dissociation was that of lead dioxide (PbO2) at 350°C. The red lead oxide known as mini-um was prepared by reacting yellow lead oxide (massicot, PbO) with air at 500°C, for a long time. In this reaction PbO become oxidized to Pb3O4 (minium). According to Debray, lead dioxide heated at 4400C and atmospheric pressure, was first rapidly destroyed, and then more slowly. The reaction ceased after 4-5 hours when the lead had transformed into minium, Pb3O4. Hence, the oxidation of massicot in air or pure oxygen at 440°C or higher, could not generate a produce more oxidized than minium because any such material would have a dissociation pressure higher than the atmospheric one. Lead dioxide, heated to 350°C, also first decomposed rapidly, and then slower. At the end of the operation, the substance had transformed into a mixture of neutral lead oxide plumbate, (PbO, PbO), a brown green powder, having a composition intermediate between minium and lead dioxide. This product was also formed when a stream of oxygen or air, at atmospheric pressure, was passed over PbO or lead carbonate heated to 350°C. The transformation of PbO into Pb3O4 was never complete; the latter transformed slowly at 350° and rapidly at 440°C, into minium, which was unable to further oxidize in air or pure oxygen, According to Debray, this phenomenon was similar to that observed with iron sesquioxide, which at a high temperature transformed into the magnetic oxide but was unable to oxidize further at any temperature and reproduce the sesquioxide (Debray, 1878).
Debray repeated his assertion that the necessary condition for a component formed directly to be able to decompose partially at a given temperature, was that its elements, after being separated by heat, be able to recombine again. Although this phenomenon was very common, in certain cases the influence of heat could modify the state of a body and leave it inadequate for assuming the combinations that it was able to assume under different conditions (e.g. what occurred during the dissociation of mercury oxide when left to cool, see above).
Within the same subject, Deville and Debray were also able to explain William Robert Grove’s (1811-1896) result that incandescent platinum was able to decompose water into an explosive mixture of hydrogen and oxygen. Grove believed that this phenomenon was an effect of heat and not of electrolysis or from that obscure mode of action called catalysis (Grove, 1847). According to Deville and Debray, water decomposed not by a mysterious property of platinum but simply because it was at a temperature well below that of its formation (about 2500°C) and that of the fusion of platinum (between 1800° and 2000°C) (Deville, 1863).