





La hipertrigliceridemia (HTG) afecta del 15 al 20% de la población mundial, y frecuentemente se descubre como un hallazgo incidental en un examen de laboratorio. Las alteraciones del metabolismo de los triglicéridos (TG) presentan bases genéticas muy complejas. Las nuevas herramientas genéticas que permiten un abordaje más preciso de las enfermedades han permitido redefinir la HTG y modificar su clasificación englobándola en dos grupos: las HTG monogénicas (TG >10 mmol/L) y las poligénicas (2 mmol/L< TG< 10 mmol/L) con un fenotipo más leve, pero con una clara influencia genética. En aproximadamente el 50% de pacientes que presentan HTG severa todavía no se ha encontrado la causa genética. Aparte de la inclusión de cada vez más genes a estudio, actualmente también se están considerando modelos estadísticos que contemplen interacciones complejas de genes-ambiente que pudieran explicar que la presencia de un conjunto de variantes aparentemente benignas pueda causar HTG ante la aparición de un factor desencadenante como la adiposidad.
El conocimiento de la naturaleza genética de las HTG también ha permitido identificar dianas para los tratamientos farmacológicos, evitando la dieta estricta con un contenido graso inferior al 20% que es complicada de mantener. El diagnóstico adecuado de estas alteraciones es fundamental para un correcto tratamiento en función del riesgo inherente a cada HTG, ya que, tal y como se ha evidenciado en múltiples estudios la concentración elevada de TG en ayunas y en estado postprandial son un factor de riesgo independiente de enfermedad cardiovascular.
Diagnosis and treatment of triglyceride metabolism disorders: from pathophysiology to clinical practice. Hypertriglyceridaemia (HTG) affects 15%-20% of the world's population, and is frequently discovered as an incidental finding in a laboratory test. Disorders of triglyceride (TG) metabolism have a complex genetic basis. New genetic tools that allow a more precise approach to the disorders have made it possible to redefine and classify HTG into two groups: monogenic HTG (TG>10 mmol/L) and polygenic HTG (2 mmol/L
Las bases genéticas de las hipertrigliceridemias (HTG) son de etiología compleja. Sin embargo, la implementación de técnicas de secuenciación masiva de forma progresiva en los laboratorios clínicos ha permitido un gran avance en los conocimientos de los genes implicados. El término primaria se refiere a aquellas HTG que tienen una base genética ya sea monogénica o poligénica, evitando de esta forma el término empleado tradicionalmente familiar, ya que sugería que la responsabilidad recaía en un gen único, es decir, se refería a las HTG monogénicas1. Aunque la HTG primaria generalmente no es consecuencia del defecto en un solo gen que muestra una herencia claramente mendeliana, por lo general, tiene una base genética, aunque es de naturaleza más compleja.
La HTG primaria es un trastorno hereditario del metabolismo lipídico caracterizado por un aumento de la concentración plasmática de triglicéridos (TG) y es una alteración relativamente frecuente. Su prevalencia estimada está en torno al 0,5 al 1% de la población general y del 5% entre los supervivientes de infarto de miocardio2. Se considera diagnóstico de HTG una concentración plasmática de TG en ayunas superior a 2,0 mmol/L (175 mg/dL)3. El panel de consenso de la Sociedad Europea de Aterosclerosis considera la HTG leve o moderada cuando la concentración de TG se encuentra entre 2,0 a 10,0 mmol/L (175 a 885 mg/dL) y grave cuando supera los 10,0 mmol/L (>885 mg/dL)4. La expresión fenotípica de la HTG primaria suele ser fenotipo IV (caracterizado por la elevación de lipoproteínas de muy baja densidad [VLDL]) y más raramente fenotipo V (con elevación de VLDL y quilomicrones [Qm]) de la clasificación de la Organización Mundial de la Salud (OMS).
Las HTG primarias son formas genéticas, monogénicas o poligénicas, donde se puede observar agregación familiar y cuya expresión puede verse modulada por la coexistencia de determinados factores higiénico-dietéticos presentes en el individuo.
Además de la HTG primaria con agregación familiar, pueden existir individuos con HTG en los que, tras realizar el estudio familiar, la concentración de TG sea inferior a 2,0 mmol/L (175 mg/dL) y no se detecte ninguna enfermedad o situación responsable de HTG. Es posible que el sujeto presente una HTG aislada o asociada a un incremento de colesterol, originándose una hiperlipidemia mixta cuya etiología puede compartir una base genética o coexistir como patologías independientes como una hipercolesterolemia poligénica o una HTG secundaria.
La primera clasificación de las hiperlipoproteinemias realizada por Fredrickson3, basada en el patrón electroforético de las lipoproteínas incluía en su definición el término HTG en cinco de los seis fenotipos descritos para su clasificación. Los diferentes fenotipos asociados a HTG están definidos por las diferentes lipoproteínas ricas en TG acumuladas, incluyendo los Qm, las VLDL y sus remanentes. En este sistema de clasificación está implícita la idea de que las diferencias entre los fenotipos asociados a HTG son debidas a variaciones genéticas, sin embargo, en la actualidad, parece que no es tan obvio porque hay un amplio solapamiento entre fenotipos causados por el mismo trastorno genético4-8.
La utilidad de este sistema de clasificación es limitada, ya que no ha mostrado un mejor conocimiento de las bases de las HTG ni tampoco ha sido útil para el tratamiento o la predicción de eventos. Por ello, se propuso que la concentración de TG junto con la presencia de otros factores de riesgo deben ser los que dirijan el manejo clínico de los pacientes con HTG1.
La era genómica ha proporcionado un nuevo paradigma para varias enfermedades genéticas y las HTG no son una excepción. Las clasificaciones tradicionales más empleadas para las hiperlipoproteinemias primarias se han basado en que la mayoría de los trastornos eran monogénicos, ya fueran dominantes o recesivos9. Sin embargo, la mayoría de dislipidemias, especialmente aquellas caracterizadas por una HTG grave, son en realidad enfermedades mucho más complejas8–10. Sin ir más lejos, las dos HTG primarias más comunes, la hiperlipidemia familiar combinada (HFC) y la hipertrigliceridemia familiar (HTGF) tienen una considerable superposición clínica y acumulan las mismas variantes genéticas comunes con pequeños efectos sobre los TG8.
El documento de consenso de la Sociedad Europea de Ateriosclerosis, clasifica las HTG primarias en tan solo dos grupos, basándose en el conocimiento de la nueva era genómica; las HTG monogénicas y las HTG multigénicas (o poligénicas).
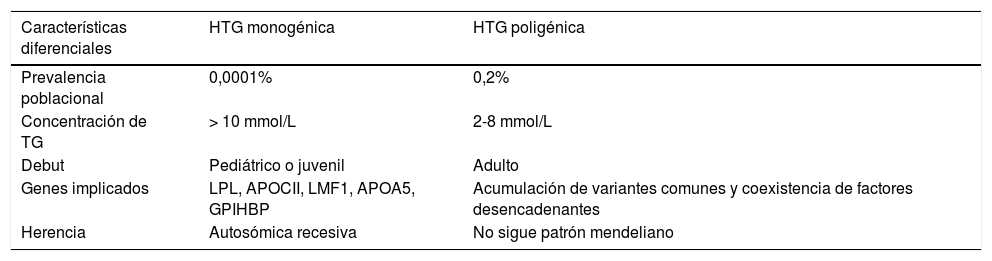
HTG monogénicas: Las HTG monogénicas son un grupo raro de enfermedades autosómicas recesivas con una prevalencia del 0,0001% en la población general. Los individuos presentan una concentración plasmática de TG superior a 10 mmol/L y su presentación tiene lugar en la edad pediátrica o juvenil y episodios de pancreatitis1. Estos individuos son homocigotos o heterocigotos compuestos para las mutaciones de pérdida de función de la lipoproteína lipasa (LPL), la apolipoproteína CII (APOCII), el lipase maturation factor 1 (LMF1), la apolipoproteína A5 (APOA5) y la proteína «glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1» (GPIHBP).
HTG multigénicas: La HTG multigénica es consecuencia de una sobrecarga de variantes genéticas comunes de pequeño efecto individual sobre los TG11. A la vez, puede haber contribución de variantes en heterocigosidad de gran efecto en genes asociados directa o indirectamente con la concentración plasmática de TG. La enfermedad se manifiesta frecuentemente a partir de la segunda década de la vida, y en la mayor parte de los casos es asintomática, basándose el diagnóstico en el hallazgo de HTG. Presentan, por tanto, un fenotipo más leve con una clara influencia genética, pero sin un efecto potente solo de un gen (tabla 1). Este subgrupo incluye la mayoría de las situaciones (>99%)1. Los individuos con HTG multigénica poseen variaciones frecuentes en genes con efectos pequeños hipertrigliceridemiantes y cuya combinación es la responsable de la HTG4–12. Conviene recordar que los TG de la dieta son transportados en plasma por los Qm, mientras que los TG sintetizados en el hígado lo son por las VLDL. Las consecuencias clínicas de la elevación de un tipo u otro de lipoproteína y de sus partículas remanentes son muy distintas, por lo que es importante el diagnóstico correcto para el adecuado tratamiento dietético y farmacológico del paciente13. De hecho, la elevación de la concentración plasmática de TG como factor de riesgo de enfermedad cardiovascular ha sido objeto de controversia14. Sin embargo, existen múltiples evidencias que abarcan estudios epidemiológicos, observacionales, ensayos clínicos de intervención e, incluso, de aleatorización mendeliana que demuestran que los TG, tanto medidos en estado de ayuno o en el postprandial, son un factor de riesgo independiente de enfermedad cardiovascular15–20. Existe otra clasificación de las HTG primarias según su riesgo cardiovascular, diferenciándose en: HTG con riesgo cardiovascular muy aumentado (HFC, disbetalipoproteinemia, HTG con dislipidemia aterogénica), HTG con riesgo cardiovascular probablemente aumentado (HTGF) y HTG sin aumento del riesgo cardiovascular (quilomicronemias primarias).
Principales características diferenciales entre las hipertrigliceridemias monogénicas y las poligénicas
| Características diferenciales | HTG monogénica | HTG poligénica |
|---|---|---|
| Prevalencia poblacional | 0,0001% | 0,2% |
| Concentración de TG | > 10 mmol/L | 2-8 mmol/L |
| Debut | Pediátrico o juvenil | Adulto |
| Genes implicados | LPL, APOCII, LMF1, APOA5, GPIHBP | Acumulación de variantes comunes y coexistencia de factores desencadenantes |
| Herencia | Autosómica recesiva | No sigue patrón mendeliano |
APOA5: apolipoproteína A5; APOCII: apolipoproteína CII; GPIHBP: glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1; HTG: hipertrigliceridemia; LMF1: lipase maturation factor 1; LPL: lipoproteína lipasa; TG: triglicéridos.
Existen estudios que han secuenciado genes en individuos con concentraciones de TG superiores al percentil 95 y de personas con concentraciones de TG en el intervalo de referencia. Los individuos con HTG tienen una frecuencia de variantes alélicas de susceptibilidad a la HTG que oscila entre dos y cinco veces más alta que aquellos con concentraciones de TG normales7. Se sabe con certeza que estas variantes son causales de HTG; sin embargo, no se asocian de forma individual con la HTG e incluso se pueden encontrar en individuos normolipidémicos. Esto nos debe incitar a reflexionar a que la HTG no posee un rasgo de herencia mendeliana en la mayoría de familias con un paciente hipertrigliceridémico.
Los estudios de asociación de genoma completo (GWAS) muestran que la HTG se relaciona de forma potente con varios genes tales como APOA5, GCKR, LPL, y APOB21. El Global Lipid Genetic Consortium ha identificado un total de 32 genes que están asociados con HTG y estos mismos loci se encuentran vinculados con pequeñas variaciones de TG dentro de los intervalos considerados normales de los mismos en población sana21. La mayoría de las HTG primarias son multigénicas (poligénicas) y están originadas por la acumulación de pequeños efectos de variantes frecuentes y efectos de variantes raras en heterocigosidad con efecto profundo sobre los TG. Es improbable que una variante común o rara de forma individual sea suficiente para causar una HTG, ya que se requieren efectos adicionales de interacción, gen-gen y gen ambiente para la expresión de la HTG.
Hay que tener en cuenta que existen otras dislipemias que cursan con HTG, pero no en exclusiva, y que tienen un origen genético, cuyo conocimiento permite su diagnóstico y su tratamiento adecuado. Se encuentran la disbetalipoproteinemia y las quilomicronemias primarias.
El diagnóstico de la disbetalipoproteinemia es muy importante porque se produce una acumulación de lipoproteínas remanentes muy aterogénicas ya que existe una presencia anormal de partículas β-VLDL circulantes. Estas hacen referencia a los residuos de Qm y VLDL (IDL), cuya movilidad electroforética se produce en la banda β (propia de las lipoproteínas de baja densidad [LDL] en lugar de la pre-β (característica de las VLDL normales). El proceso arterioesclerótico se acelera en las formas hiperlipémicas, ocasionando un fenotipo de hiperlipemia tipo III que se caracteriza por un aumento de las concentraciones plasmáticas de TG y colesterol (entre 300 y 1.000 mg/dL). La prevalencia es baja (0,1 a 0,4%) pero es posible que esté infradiagnosticada22. Se manifiesta en la adolescencia o después de los 20 años y normalmente necesita un factor desencadenante. En su patogenia, es clave la APOE, que es una glucoproteína de 34 KDa de 299 aminoácidos que constituye el elemento de reconocimiento de dichas partículas residuales por sus receptores hepáticos23. El gen que la codifica está en el cromosoma 19 (19q13.2), es polimórfico y contiene cuatro exones. Las tres variantes principales constituyen los alelos E3, E4 y E2 (por orden de frecuencia), que se diferencian entre sí por los aminoácidos de dos posiciones de la proteína (cisteína o arginina en las posiciones 112 y 158 de la proteína). Las combinaciones de estos tres alelos dan lugar a los seis genotipos más habituales24. El genotipo E3/E3 es el más frecuente y no está asociado a disbetalipoproteinemia, el genotipo E3/E2 posee cierta predisposición a desarrollar la enfermedad, el genotipo E3/E4, que no se relaciona con disbetalipoproteinemia y genotipo E4/E4, asociado a hipercolesterolemia y a enfermedad de Alzheimer. Finalmente es el genotipo E2/E2 el que está vinculado a la disbetalipoproteinemia. Los homocigotos E2/E2 tienen mayor dificultad para aclarar estas partículas residuales por el hígado y se produce su acumulación en el plasma. Este genotipo tiene una frecuencia en la población del 1%, pero solo desarrolla la enfermedad uno de cada 50 a 100 homocigotos. Esto sugiere una base genética compleja también. Para que haya expresión de la enfermedad deben existir otras alteraciones genéticas o enfermedades (obesidad, diabetes, hipotiroidismo) o ambientales (consumo de alcohol o ciertos fármacos). En estas circunstancias, se reduce la actividad de los receptores hepáticos o se saturan, al estar incrementada la síntesis de VLDL y colesterol por el hígado. La disbetalipoproteinemia asociada al genotipo E2/E2 se comporta como una enfermedad hereditaria de tipo recesivo y se manifiesta en la vida adulta, con predominio en hombres hasta la edad de la menopausia. Existen otras variantes menos comunes de la APOE (infrecuentes) que pueden originar casos raros25 de disbetalipoproteinemia con patrón de herencia dominante y posible presentación infantil. Incluso, puede aparecer en heterocigotos sin necesitar la presencia de ningún otro factor condicionante. En general las mutaciones en APOE se transmiten de forma autosómica recesiva, pero en el 10% de los casos de forma dominante22. Hasta la fecha, se han descrito aproximadamente más de 30 variantes de APOE de las cuales 14 se asocian a disbetalipoproteinemia y otras siete a otras hiperlipemias. Ante una sospecha de disbetaliproteinemia es útil estudiar si está presente el genotipo E2/E2, sin embargo, esto ha provocado que no se secuencia de forma completa el exón 4 del gen APOE, claramente polimórfico, y ha podido provocar que las disbetalipoproteinemias no E2/E2 no se detectasen.
La sospecha clínica de disbetalipoproteinemia surge ante un perfil lipídico y su confirmación se basa o bien en la existencia del genotipo E2/E2 del gen APOE (en el contexto clínico adecuado) o en la detección las VLDL mediante ultracentrifugación de las lipoproteínas plasmáticas y/o un cociente entre el colesterol ligado a VLDL dividido por los TG totales superior a 0,3. Actualmente, se proponen el empleo de cocientes como APOB/no lipoproteínas de alta densidad (HDL) para ayudar en el cribado de la enfermedad y seleccionar aquellos individuos sospechosos de disbetalipoproteinemia para realizar posteriormente el estudio genético o la ultracentrifugación26.
En el caso de las quilomicronemias primarias, se observa un fenotipo de HTG con Qm. Ocurre algo similar a lo comentado en el caso de las HTG primarias. Hasta hace muy poco, la referencia a la quilomicronemia (QM) familiar incluía solo tres trastornos hereditarios: el déficit familiar de LPL, déficit familiar de APOCII y el inhibidor familiar de LPL. En los últimos años se han incluido otras mutaciones en genes que codifican proteínas que modulan la actividad de la LPL y que causan hiperquilomicronemia (HQM). Esto ha producido un cambio en la clasificación y definición de estos trastornos.
Las HQM se presentan en dos formas primarias distintas: la quilomicronemia familiar (o hiperlipoproteinemia tipo I), monogénica, rara, de aparición en la infancia o en la adolescencia27; y la quilomicronemia poligénica (hiperlipoproteinemia tipo V), de aparición tardía, causada por la acumulación de variantes genéticas, y que se pone de manifiesto cuando concurren factores secundarios tales como dieta inadecuada, obesidad, ingesta excesiva de alcohol, o diabetes mellitus mal controlada. Es más frecuente que la forma familiar monogénica1.
La HQM primaria es una dislipemia hereditaria autosómica recesiva, caracterizada por la acumulación de Qm en el plasma en ayunas. La causa es un defecto en el catabolismo de los TG transportados por los Qm, debido al déficit de actividad de la enzima LPL o bien a mutaciones en los genes que codifican proteínas que afectan a su actividad. Desde hace ya mucho tiempo, se habían descrito mutación en los genes LPL y APOC2 (cofactor necesario para que la LPL funcione correctamente). El complejo enzimático de la LPL cataliza la hidrólisis de los enlaces 1 y 3 de los TG del núcleo de los Qm y de las VLDL circulantes, formando monoglicéridos, diglicéridos y ácidos grasos libres. Los ácidos grasos libres serán captados por los tejidos para su utilización (oxidación) o su almacenamiento (tejido adiposo). En condiciones normales este proceso se completa en pocas horas tras la ingesta. Cuando tiene lugar una HQM primaria este proceso se ve alterado. En la forma monogénica los defectos bioquímicos consisten en un déficit de la LPL, de su cofactor APOCII, o de otras proteínas implicadas en su actividad. Las más importantes definidas hasta ahora son la apolipoproteína A-V (APO A-V), el factor de maduración de las lipasas (LMF1), también denominada chaperona de la LPL, y la proteína de unión a HDL tipo 1 anclada por glicofosfatidilinositol (GP1HBP1). Cada uno de ellos son fundamentales para el correcto funcionamiento de la LPL. Mutaciones graves heredadas en homocigosis o en heterocigosis compuestas, de alguno de estos cuatro genes, impiden el correcto funcionamiento de la LPL, dando lugar a una HTG grave caracterizada por el acúmulo de Qm en plasma durante el ayuno28. En los pacientes afectados no hay evidencia de aumento del riesgo de padecer una enfermedad cardiovascular prematura, ya que los Qm no son partículas aterogénicas. Sin embargo, los pacientes suelen tener retraso en el desarrollo, además de un importante riesgo de presentar episodios recidivantes de pancreatitis aguda, pudiendo finalmente dar lugar a una insuficiencia pancreática crónica. Por ello, es importante el diagnóstico y tratamiento precoz con restricción estricta de grasa dietética, o nuevos fármacos como los nuevos oligonucleótidos antisentido de segunda generación que se dirige al ARN mensajero para la APOC3 para evitar las complicaciones posteriores y mejorar la calidad de vida de estos pacientes29,30.
La segunda forma de quilomicronemia, la poligénica, se caracteriza por tener una expresión tardía, junto con una reducción de la concentración en plasma de colesterol de HDL7. Además, se observan una gran cantidad de disfuncionalidades lipídicas, que incluyen el aumento de partículas remanentes de VLDL, lipoproteínas remanentes con APOB-100, y descenso de la concentración de colesterol de HDL31. En estos casos, la hiperquilomicronemia constituye un marcador de lipemia postprandial que se considera un estado metabólico preaterogénico32. Resultan especialmente proaterogénicos los remanentes de lipoproteínas ricas en TG que se acumulan tras la ingesta, tanto los que son de origen intestinal, que contienen APOB-48, como los de origen hepático, que contienen APOB-100, aunque hay que señalar que resulta complicada la medición de la concentración de colesterol de estas lipoproteínas en la práctica clínica mediante procedimientos estandarizados.
La nueva era de la medicina genómica ha cambiado el paradigma de las enfermedades, incluyendo las dislipemias. Ha sido necesaria una nueva clasificación y definición de las HTG tras el conocimiento de sus bases genéticas que está en continua evolución y que debemos ir adaptando a la práctica clínica habitual.
Conflicto de interesesLos autores declaran que no tienen conflicto de intereses.
Clínica e Investigación en Arteriosclerosis. Número monográfico.
Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica.
- Documento de recomendaciones de la Sociedad Española de Arteriosclerosis (SEA). La dieta en la prevención cardiovascular. Actualización 2024
- “Notas metodológicas”: una nueva e importante sección en Clínica e Investigación en Arteriosclerosis
- Consenso sobre lipoproteína (a) de la Sociedad Española de Arteriosclerosis. Revisión bibliográfica y recomendaciones para la práctica clínica
recomendados