











El objetivo del presente estudio fue identificar los efectos de la transfección in vivo del ARNsi de MCP-1 en un modelo de aterosclerosis sobre la expresión local de MCP-1 y la patogenia de la enfermedad.
MétodosEn 28 conejos blancos de Nueva Zelanda, se indujo aterosclerosis carotídea. Los animales se dividieron aleatoriamente en tres grupos: grupo ARNi, grupo modelo y grupo de plásmido blanco. El vector de expresión ARNsi se transfectó a los vasos sanguíneos a través de liposomas. Las arterias carótidas se procesaron para su evaluación morfológica. La expresión local de MCP-1 se detectó mediante inmunohistoquímica, reacción en cadena de la polimerasa retrotranscriptasa e inmunoelectrotransferencia.
ResultadosEn las secciones teñidas con hematoxilina y eosina, en el grupo ARNi, comparado con el grupo modelo y el grupo plásmido blanco, se observó desprendimiento de las células endoteliales, al mismo tiempo que una menor hiperplasia íntimal. El cociente I:M disminuyó significativamente hasta 1,45 en el grupo ARNi comparado con el grupo modelo y el grupo plásmido blanco (5,55 y 5,27, respectivamente). Los resultados de la inmunohistoquímica demostraron que la expresión de MCP-1 fue menos intensa en el grupo ARNi. La RCP-RT y la inmunoelectrotransferencia demostraron una disminución de la expresión en el grupo ARNi comparado con el grupo modelo y el grupo plásmido blanco. Se detectaron correlaciones muy positivas entre la RCP-RT semicuantitativa y el cociente I:M (r=0,968).
ConclusiónLa expresión de MCP-1 se inhibió satisfactoriamente mediante la transfección del plásmido de expresión del ARNsi MCP-1 en la arteria carótida, y la progresión de la aterosclerosis se limitó mediante el silenciamiento de la expresión de MCP-1 mediada por ARNsi.
La estenosis de la arteria carótida es una importante amenaza para la salud, que da lugar a discapacidad y muerte, siendo una de las causas principales de ictus1. Durante más de medio siglo se han estudiado los mecanismos que son la base del desarrollo de la aterosclerosis (AS). Para explicar la aterogénesis, se han formulado tres principales teorías, a saber, la de la infiltración lipídica; la de la lesión y la corrección; así como la de la trombosis. A medida que los estudios anatomopatológicos experimentales han avanzado, los investigadores han descubierto que las placas de AS no sólo muestran una acumulación sustancial de lípidos sino también la infiltración de un elevado número de células inflamatorias y secreción de citocinas inflamatorias. En 1976, Ross y Glomset2 propusieron la teoría de “la lesión-corrección” para explicar la formación de estas placas. En la década de los ochenta, muchos investigadores efectuaron numerosos estudios relacionados con los factores inflamatorios, marcadores inflamatorios y placas de AS y consideraban que, en su desarrollo y progresión, participaba la respuesta inflamatoria. En 1999, Ross propuso explícitamente que la AS era un tipo de enfermedad proliferativa inflamatoria. Consideraba que la formación de las lesiones de AS era consecuencia de la fibroplasia inflamatoria de la arteria tras la respuesta a la disfunción endotelial arterial, causada por diversos factores de riesgo mayores de esta enfermedad3-7. En la AS los primeros cambios morfoanatomopatológicos se caracterizan por la migración de los monocitos/macrófagos en la túnica intimal y la formación de las células espumosas subendoteliales, seguido de la migración (desde la túnica media a la túnica íntima) e hiperplasia de las células musculares lisas (CML). En la actualidad, en general se considera que la migración de los monocitos en el endotelio arterial, mediada por la proteína 1 quimiotáctica monocítica (MCP-1), desempeña un papel clave en el desarrollo precoz de esta enfermedad. Como miembro de la subfamilia de las quimiocinas CC, la MCP-1 es una glucoproteína con un peso molecular de 14 x 103. Los estudios revelan que las células endoteliales vasculares (CEV), las células musculares lisas vasculares (CMLV), monocitos, macrófagos (Mφ) y fibroblastos expresan MCP-1. A través de una acción específica en los monocitos de sangre periférica y con su atracción para que migren en el subendotelio, la MCP-1 desempeña un importante papel en el desarrollo y progresión de la AS8,9. Yla-Herttula et al10 examinaron la expresión de esta proteína en lesiones de AS en conejos y seres humanos utilizando inmunohibridación, hibridación in situ e inmunohistoquímica. Encontraron que la MCP-1 se expresaba positivamente en arterias AS de conejo y lesiones de AS ricas en células espumosas, derivadas de Mφ, aisladas de macrófagos. En comparación, la MCP-1 se expresó negativamente en arterias sanas y en Mφ alveolares aislados del mismo animal. En la hibridación in situ se demostró que el ARNm de MCP-1 se expresaba positivamente en las lesiones AS de conejo ricas en macrófagos, mientras que el análisis inmunohistoquímico demostró adicionalmente que la proteína se expresaba en las regiones positivas para ARNm MCP-1. En estas regiones, también se expresaban positivamente las lipoproteínas de baja densidad oxidadas (ox-LDL), lo que concuerda con la observación de que éstas podrían inducir la expresión in vitro de MCP-1 en las CEV y las CMLV, lo que sugiere que las ox-LDL podrían participar en el desarrollo de las lesiones de AS induciendo la expresión de la proteína. A través del estudio de la túnica intimal de la arteria carótida de pacientes sometidos a endarterectomía y comparándola con la de individuos con arterias sanas, Nelken et al11 encontraron que la MCP-1 se tiñó positivamente en el 16% de las células de arterias carótidas AS, mientras que el porcentaje de células MCP-1 positivas en arterias sanas fue<0,1%, lo que demuestra que la proteína desempeña un papel en la migración de los monocitos en la pared de las arterias AS. Schwartz et al12 demostraron que la MCP-1 es un factor quimiotáctico monocítico potente que puede regular la expresión de las moléculas de adhesión pertinentes en el endotelio vascular. Después del cruzamiento de ratones con una deleción de CCR2 (un receptor de MCP-1) (CCR2 -/-) con ratones con una deleción de apolipoproteína E (apoE) (apoE -/-), Boring et al13 descubrieron que la deleción de CCR2 pudo reducir significativamente la incidencia de AS. Estudios similares también demostraron que en ratones apoE -/- la aceleración de la AS inducida por la sobreexpresión de MCP-1 se asociaba con un aumento del número de macrófagos en las placas de AS14-16. Estos descubrimientos indican que la MCP-1 desempeña un importante papel en la formación de la AS, y se ha reconocido como una nueva diana para su prevención y tratamiento.
Los investigadores tanto nacionales como extranjeros han tratado de examinar la posibilidad de usar la MCP-1 como diana del tratamiento antiinflamatorio17-20. En teoría, para inhibir la expresión de los genes diana, pueden usarse numerosas tecnologías, como la tecnología antisentido, tecnología de ribozima, al igual que tecnologías basadas en ácidos nucleicos y péptidos de pequeña molécula. No obstante, estas moléculas o fármacos no se diseñan fácilmente. Además, sólo es posible obtener las moléculas que pueden suprimir específica y eficientemente la expresión de los genes diana después de efectuar múltiples pruebas de ensayo y error y exámenes de cribado. La aparición de la tecnología de interferencia del ARN (ARNi) proporciona un nuevo indicio para bloquear la expresión de esta proteína. Como nueva tecnología para inactivar los genes diana, la ARNi exhibe las ventajas de un diseño más sencillo, al igual que una mayor eficiencia de inhibidores y especificidad comparada con las tecnologías mencionadas previamente. Por esta razón, ha desempeñado importantes papeles en el estudio de la función génica y el cribado de dianas de los fármacos en los últimos años21-24. En la actualidad, en la ARNi se usan principalmente dos estrategias. Una es la transfección directa de pequeños ARN de interferencia (si) (sintetizados químicamente o transcritos in vitro) en las células; la otra es introducir un sistema de vector (plásmido o vectores víricos) en las células que pueden expresar endógenamente ARNsi.
En el presente estudio construimos un vector de expresión eucariótico de ARNsi específico de MCP-1 y establecimos un modelo de AS carotídea de conejo. Después de la transfección local mediada por liposomas, observamos la influencia del ARNsi MCP-1 en la expresión de la proteína en la arteria y analizamos adicionalmente el efecto de ARNsi MCP-1 sobre la AS. Los resultados proporcionarán nuevos indicios para el desarrollo de estrategias intervencionistas novedosas de prevención y tratamiento de esta enfermedad.
Materiales y métodosConstrucción del vector de ARNsi MCP-1El vector pSilencer 2.0-U6 se adquirió a partir de Ambion (Austin, TX). La cepa DH-5a de Escherichia coli fue proporcionada por el Department of Biochemistry de nuestra universidad (Guangzhou, China). Las endonucleasas de restricción BamHI e HindIII, T4 DNA ligasa y ampicilina se adquirieron a partir de Promega (Madison, WI). El estándar de peso molecular de ADN (λ-EcoT14Idigest) se adquirió a partir de Beijing Ding-guo Biotechnology (Beijing, China). Los equipos de purificación y recuperación de ADN se obtuvieron a partir de Beijing JingshiBio-engineering (Beijing, China). Para la preparación de ADN de plásmido a pequeña escala se utilizaron equipos Qiagen (Chatsworth, CA).
Síntesis de ADNdsEn función de la secuencia del gen de MCP-1 depositada en GenBank se designaron dos pares de ARNsi. Los correspondientes ADN de doble cadena (double stranded [ds]) fueron sintetizados por Shanghai Invitrogen Biotechnology (Shanghái, China), designados como MCP:sh1 y MCP:sh2. Tras la disolución del ADNds en el tampón TE, se mezclaron 5μl de la solución de cada ADN de cadena simple para perimitir la anelización (2min a 95°C, 2min a 72°C, 2min a 37°C, 2min a 25°C, y 10min en un baño de hielo) para formar ADNds para el futuro ligamiento.
sh1:
5'-GATCCGCTGCTATAACTTCACCAATT-CAAGAGATTGGTGAAGTTATAG-CAGCTTTTTGGAAA-3'
5'-AGCTTTTCCAAAAAGCTGCTATAACTT-CACCAATCTCTTGAATTGGTGAAGTTATAG-CAGCG-3'
sh2:
5'-GATCCGCTGTGATCTTCAAGACCATT-CAAGAGATGGTCTTGAAGATCACAGCTTTTTGGAAA-3'
5'-AGCTTTTCCAAAAAGCTGTGATCTTCAA-GACCATCTCTTGAATGGTCTTGAAGATCA-CAGCG-3'
Las letras en negrita representan las cadenas sentido y antisentido de la secuencia diana a interferir.
Doble digestión del vector y recuperación del vector digeridoEn función del mapa estructural del plásmido psilencer2.0-U6 (Fig. 1), se digirió doblemente dicho plásmido con BamHI e HindIII (en un volumen total de reacción de 20μl, que consistió en 10μl de plásmido, 2μl de 10 x tampón K, 1μl de BamHI, 1μl de HindIII, y 6μl de agua doble destilada; incubación a 37°C durante 4h). De acuerdo con las instrucciones suministradas con el equipo de purificación y recuperación de ADN, los productos digeridos se resolvieron mediante electroforesis en 1% de gel de agarosa, recuperándose el fragmento psilencer2.0-U6 linealizado.
Ligamiento del templado de ARNsi en el vector
En el sistema de reacción de ligamiento, el cociente molar del templado de ARNsi con respecto al vector fue de 3-5:1 (en un volumen de reacción total de 15μl, que contenía 1,5μl del tampón T4 ADN, 1μl de T4 ADN ligasa y 8,5μl de agua doble destilada). Después de mezclarlo completamente, el sistema de la reacción se incubó a 37°C durante 4h. Los productos ligados pudieron transformarse de inmediato en las células competentes o almacenarse a –20°C.
TransformaciónLos productos ligados se transformaron en células competentes (preparadas de acuerdo con el método del cloruro cálcico descrito en el manual “Clonación molecular”). Los procedimientos detallados de transformación fueron los siguientes: los productos ligados se mezclaron en un baño de hielo durante 30min, y se incubaron en un baño a 42°C (estrés térmico [heat-shock]) durante 90 s, y se introdujeron de nuevo en el baño de hielo durante 1-2min después de añadir 1ml de medio de Luria-Bertani (LB), la mezcla se incubó en una agitadora a 37°C durante 1-2h. El cultivo resultante se sembró y cultivó a 37°C durante 12-16h. Una colonia individual se introdujo en 5ml de medio LB y se cultivó toda la noche. Acto seguido, se condujo el aislamiento del plásmido a pequeña escala utilizando el equipo anteriormente referido. Tras la identificación por doble digestión con BamHI e HindIII, se remitieron los plásmidos identificados para su secuenciación.
Preparación a gran escala del plásmido recombinanteDespués de la secuenciación, se transformó el plásmido que contenía el inserto esperado en las células competentes (utilizando el mismo método descrito previamente). La preparación a gran escala del plásmido recombinante se efectuó utilizando el método de lisis alcalina descrito en el libro “Clonación molecular”. La concentración de la solución del ADN de plásmido purificado se determinó utilizando un espectrofotómetro.
Transfección local de ARNsi de MCP-1Principales reactivosConstruimos nosotros mismos el ARNsi Psilencer2.0-U6-MCP-1; se adquirió Lipofectamine2000 a partir de Invitrogen (Carlsbad, CA); el anticuerpo murino monoclonal anti-MCP-1 se adquirió a partir de R&D Systems (Mineápolis, MN); el equipo de extracto de Trizol se adquirió a partir de Invitrogen; el equipo de retrotranscripción se adquirió a partir de Promega; el equipo de reacción en cadena de la polimerasa (RCP) se adquirió a partir de Takara Bio (Otsu, Shiga, Japón); los cebadores (primers) retrógrados y anterógrados de MCP-1 se adquirieron a partir de Invitrogen; el anticuerpo monoclonal antitubulina se adquirió a partir de Santa Cruz Biotechnology (Santa Cruz, CA).
Establecimiento del modelo animal y transfección in vivoA partir de la Guangzhou University of Traditional Chinese Medicine (permiso SYXK[GD] 2007-0081) se adquirieron 28 conejos blancos de Nueva Zelanda, 2,0-2,5kg de peso.
Se insertó un catéter montado sobre balón en la arteria carótida común a través de la arteria carótida externa izquierda. Tras lesión de la arteria carótida común inducida con balón, durante 8 semanas los conejos fueron alimentados con una dieta rica en grasas que contenía un 1% de colesterol y un 0,5% de manteca de cerdo. Por lo tanto, se obtuvo un modelo de AS en la arteria carótida común de estos animales. Los procedimientos detallados fueron los siguientes: los conejos fueron anestesiados mediante inyección intravenosa de un 3% de pentobarbital sódico en una dosis de 30mg/kg y, acto seguido, recibieron heparina en una dosis de 200 U/kg a través de la vena marginal de la oreja. Tras fijación en posición de decúbito supino, afeitado del pelo, lavado y desinfección, se efectuó un abordaje cutáneo de unos 5cm de longitud a lo largo de la línea media del cuello para exponer y separar la arteria carótida común izquierda (3,0-4,0cm). Mientras tanto, se separaron horizontalmente por encima del cartílago tiroides la arteria carótida interna y externa izquierdas. Utilizando clampajes arteriales se clamparon los extremos proximales de la arteria carótida común y de la carótida interna. Más tarde, se ligó el extremo distal de la arteria carótida externa, al mismo tiempo que el extremo proximal se hacía ascender con hilo quirúrgico para que el abordaje adoptara una forma de V (en un ángulo de unos 45°) y una localización a 0,5cm de distancia de la intersección de la arteria carótida interna y externa. Acto seguido, se procedió a la inserción retrógrada de un catéter montado sobre balón de Fogarty de 4F. Después de la inyección de una solución de suero salino fisiológico que contenía heparina para alcanzar una presión de 2 atmósferas, se hizo retroceder lentamente el catéter montado sobre balón desde el extremo proximal. Después de repetirlo tres veces, se retiró el catéter montado sobre balón. Más tarde, se ligó el extremo proximal de la arteria carótida externa izquierda y se restableció el flujo sanguíneo de la arteria carótida común izquierda y de la arteria carótida interna izquierda y se suturó la piel. Después de la cirugía, se administró una inyección intramuscular diaria de penicilina (800.000 unidades) durante 3 días consecutivos. Los conejos recibieron una alimentación rica en grasas durante 8 semanas.
Acto seguido, se asignó aleatoriamente a los animales a un grupo ARNi (n=12), un grupo modelo (n=8) y un grupo plásmido blanco (n=8). Al término de la alimentación rica en grasas durante 8 semanas, la arteria carótida común izquierda se movilizó de nuevo y los dos extremos del segmento que debía transfectarse se ocluyeron temporalmente con clips. Los transfectantes se inyectaron en el segmento ocluido con una jeringa, y la infusión se retuvo durante 30min. Los animales del grupo ARNi se transfectaron con el pSilencer2.0-U6-MCP-1-siRNA/Lipofectamine 2000, los animales del grupo modelo se transfectaron con suero salino normal, y los del grupo plásmido blanco se transfectaron con plásmido blanco/Lipofectamine 2000.
Preparación de los genesEl ARNi (50 μg) se diluyó en 250μl de suero salino normal. Acto seguido, ambos se mezclaron y se mantuvieron estáticos durante 10-15min.
En el día 7 después de la transfección, se recogieron muestras arteriales carotídeas para tinción con hematoxilina y eosina (HE) y tinción con el anticuerpo monoclonal anti-MCP-1, al mismo tiempo que algunas muestras se almacenaban a –70°C para una RCP adicional de retrotranscripción (RCP-RT) e inmunoelectrotransferencia.
Examen anatomopatológicoLas muestras se tiñeron con HE, y se tomaron fotografías con una cámara digital. Las secciones transversas de la arteria carótida se examinaron con el microscopio y morfológicamente para medir el cociente del grosor de la íntima con respecto a la media (I:M), basado en un estudio previo25, a través del programa Image Pro Plus Analysis Software (Media Cybernetics, Silver Spring, MD).
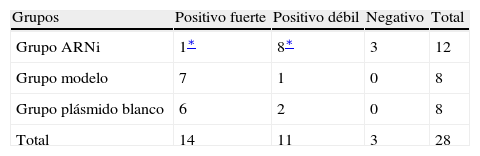
Tinción inmunohistoquímicaComo primer anticuerpo, se añadió un anticuerpo monoclonal anti-MCP-1 y la tinción se efectuó de acuerdo con las instrucciones del fabricante (Copenhague, Dinamarca) con coloración mediante diaminobencidina (DAB) y contratinción nuclear de hematoxilina. Las secciones se examinaron bajo un campo visual x 200 y se fotografiaron, mostrándose la expresión MCP-1 positiva como partículas amarillo-pardas o pardas-chocolate en el citoplasma o la membrana de las células espumosas. Dos anatomopatólogos revisaron y clasificaron todas las secciones (positiva fuerte, positiva débil y negativa).
Prueba de RCP-RTCadena sentido de MCP-1 5'-AGACCATCT-CAGTGAAGAGGCTAAT-3' y cadena antisentido 5'-AACTGGGGTTCACAGAGGGAAAGCA-3', 400 pb; cadena sentido de GAPDH 5'-AAGGCCATCACTATCTTCCA-3' y cadena antisentido 5'-CCTGCTTCAC-CACCTTCTTG-3', 579 pb. La RCP se efectuó a 94°C, con desnaturalización durante 5min a 94°C, durante 30 s a 60°C, durante 30 s a 72°C, durante 60 s, para un total de 30 ciclos, y una elongación a 72°C durante 5min. Los productos de RCP se sometieron a electroforesis en un 2% de agar, y el fotografiado se completó con un sistema de exploración de imágenes en gel. El análisis RCP-RT semicuantitativo se efectuó a través del programa Quantity One 1-D Analysis Software (versión 4.6.2) de Bio-Rad (Richmond, CA).
InmunoelectrotransferenciaSe añadió a los tejidos tampón de lisis, se homogeneizaron y se centrifugaron a 1.600rpm durante 20min. El sobrenadante se recogió para determinar la concentración de proteínas. Las muestras de proteínas (50 μg) se transfirieron a una membrana de nitrocelulosa después de electroforesis en gel de un 10% de SDS-poliacrilamida (PAGE) y bloqueada con leche en polvo desnatada; las muestras se incubaron con un anticuerpo monoclonal anti-MCP-1 durante una hora y con anticuerpos secundarios durante 30min para el desarrollo de imágenes y, acto seguido, se analizaron con un sistema de exploración de imágenes.
Análisis estadísticoTodos los análisis estadísticos se procesaron con el programa SPSS 16.0 (SPSS, Inc, Chicago, Estados Unidos). Los datos cuantitativos se expresaron como media ± desviación estándar, y se efectuó un análisis de la varianza de una vía para comparar las diferencias entre grupos. Al mismo tiempo, se efectuó una prueba de probabilidad exacta para los datos cualitativos y también se efectuó un análisis de correlación de Pearson.
ResultadosResultados de la construcción del vector de ARNsi MCP-1Construcción del plásmido psilencer2.0-U6-siMCP-1Puesto que el vector de expresión eucariótico psilencer2.0-U6 contiene un sitio de multiclonación, se usaron las endonucleasas de restricción BamHI e HindIII para linealizar el pásmido. Después del ligamiento del fragmento diana (MCP-1) en el plásmido pcDNA3 usando la T4 ADN ligasa, se obtuvo la expresión eucariótica recombinante del plásmido psilencer2.0-U6-siMCP-1 (Fig. 1).
Identificación del plásmido recombinante por doble digestiónLa inserción correcta de los fragmentos diana en los vectores de expresión es clave para la expresión de los genes exógenos. Puesto que el vector linealizado puede experimentar autoligamiento, es esencial identificar el plásmido recombinante. La doble digestión de BamHI e HindIII debería deparar dos bandas en un gel de agarosa, que representarían el plásmido de ADN (3.067 pb) y el fragmento diana (62 pb). Sin embargo, puesto que el peso molecular del fragmento diana fue mucho menor que el de plásmido de ADN e incluso insignificante, el fragmento diana tuvo una tasa de migración más rápida y migró fuera del gel. Como consecuencia, en el gel sólo apareció una banda (el plásmido de ADN). La doble digestión del plásmido recombinante indicó que el fragmento diana se había insertado direccionalmente en el vector de expresión (Figs. 2a y 2b).
Resultados de la secuenciación del plásmido recombinante positivo (psilencer2.0-U6-siMCP-1)
Los resultados de la secuenciación mostraron que la secuencia del plásmido recombinante positivo era idéntica a la del gen de MCP-1 depositado en la base de datos Genbank, lo que demuestra que el plásmido recombinante fue un vector de expresión que contenía ADNc MCP-1 (v. Figs. 3a, 3b para los resultados de la secuenciación).
Resultados de la transfección local de ARNsi MCP-1Examen anatomopatológico MCP-1: sh1. (b) MCP-1: sh2.")
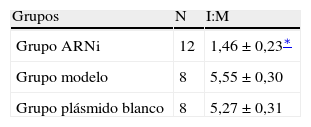
En el grupo de modelo AS y el grupo plásmido blanco, las secciones teñidas con HE de las arterias carótidas de conejo demostraron la presencia de estenosis significativa de la luz arterial, el desprendimiento de las células endoteliales, hiperplasia intimal significativa y ensanchamiento del espacio subendotelial. Así mismo, las placas consistían en células espumosas y miofibroblastos, con una distribución desordenada del músculo liso medial, la separación y rotura de la lámina elástica interna, y la penetración de las CML en la capa endotelial a través de la lámina elástica interna. En el grupo ARNi, se observó desprendimiento de las células endoteliales, al mismo tiempo que había menos hiperplasia intimal en comparación con los de los grupos modelo y plásmido blanco (Fig. 4). Microscópicamente, en el grupo ARNi el cociente I:M disminuyó significativamente en comparación con los otros dos grupos (p<0,01) (tabla I), lo que indicó menor grado de estenosis y una AS menos grave en el grupo ARNi.

Cocientes I:M de cada grupo
| Grupos | N | I:M |
| Grupo ARNi | 12 | 1,46±0,23∗ |
| Grupo modelo | 8 | 5,55±0,30 |
| Grupo plásmido blanco | 8 | 5,27±0,31 |
I:M: espesor intimal:medial.
Las secciones mostraron menor grado de tinción en el grupo ARNi y se tiñeron significativamente en los otros dos grupos, sin diferencias significativas entre ellos, lo que demostró que la transfección del vector de expresión ARNsi MCP-1 podría reducir la expresión local de la proteína (Fig. 5, tabla II).
Prueba de la RCP-RT
Como se muestra en la Fig 6, la expresión del ARNm en el grupo ARNi fue menor que en los otros dos grupos, lo que sugiere que la transfección local de ARNsi pSilencer2.0-U6-MCP-1 disminuyó la expresión de ARNm MCP-1.
Correlaciones entre la RCP-RT semicuantitativa y el cociente I:M. Línea 1, grupo ARNi; línea 2, grupo modelo; línea 3, grupo plásmido blanco.")
Se efectuó un análisis de correlación de Pearson entre la RCP-RT semicuantitativa (Fig. 7) y el cociente I:M, que reveló un r=0,968 (p<0,01) (Fig. 8) y, así mismo, correlaciones muy positivas entre ellos, con una menor expresión de MCP-1 y un menor cociente I:M.
 semicuantitativa. 1, grupo ARNi∗; 2, grupo modelo; 3, grupo plásmido blanco (∗p<0,01, comparado con los otros dos grupos).")
 y el cociente I:M (r=0,968, p<0,01). I:M: grosor intimal:medial. RTPCR: RCP-RT.")
En los grupos modelo de AS y plásmido blanco, aumentó la expresión de MCP-1 pero disminuyó tras la interferencia de ARNsi MCP-1. Ésta controló satisfactoriamente la expresión de la proteína postranscripción (Fig. 9).
Discusión
El establecimiento satisfactorio de un modelo animal de aterosclerosis desempeña un importante papel en la investigación detallada de su etiología, mecanismos, prevención y tratamiento. La reproducción de un modelo de lesión mecánica en combinación con una alimentación rica en colesterol se caracterizó por una elevada tasa de eficacia y gran similitud con la AS carotídea humana, por lo que se ha empleado con frecuencia en los últimos años26-31. El examen anatomopatológico demostró que, a las 12 semanas, en el grupo modelo las células endoteliales se desprendían con la agregación sustancial de células espumosas debajo de la capa endotelial, lo que dio lugar a una hiperplasia significativa de la íntima y a la distribución desordenada de las CML mediales, lo que coincide con las características anatomopatológicas de la AS precoz, indicando el establecimiento satisfactorio del modelo de conejo de AS carotídea mediante denudación con balón del endotelio arterial carotídeo en combinación con una dieta rica en grasas.
Tanto la RCP-RT como la inmunoelectrotransferencia confirmaron que, en los grupos tanto modelo como control, el ARNm MCP-1 y la proteína se expresaron en las arterias lesionadas; los hallazgos inmunohistoquímicos demostraron que MCP-1 se expresaba principalmente en el citoplasma y membrana de los macrófagos (principalmente, las células espumosas), CEV, y CMLV, en particular en los macrófagos y las células espumosas, lo que coincide con los estudios previos publicados por Yla-Herttula et al10 y Nelken et al11. La MCP-1 es la proteína quimiotáctica monocítica, que induce la migración de los monocitos hacia la íntima, y que es activada en los macrófagos y células espumosas después de la fagocitosis de lípidos, y desempeña un papel decisivo en la aterosclerosis precoz. Como proteína quimiotáctica monocítica sumamente potente, no sólo es capaz de adherirse a los monocitos móviles, sino también de regular la expresión de las moléculas adherentes endoteliales vasculares. Schwartz et al12 describieron que, como proteína con las características mencionadas previamente, fue capaz de regular las moléculas adherentes relacionadas con la expresión endotelial vascular, manifestando sus dobles funciones de regulación de la adherencia y migración de los monocitos en la pared arterial y desempeñando un papel decisivo en el proceso de migración de los monocitos en la íntima durante las fases iniciales de la AS. Algunos autores32,33 han descrito la inhibición de la migración de los macrófagos como una importante estrategia de prevención y tratamiento de la enfermedad, al mismo tiempo que esta proteína se ha convertido en una nueva diana para inhibir la migración de los macrófagos hacia el interior de la pared vascular.
La técnica de ARNi es capaz de inhibir de forma selectiva, efectiva y específica la expresión de los genes diana utilizando pequeños fragmentos de ARN de doble cadena, de 21-23 pb (ARNsi) para inducir la degradación del ARNm de secuencia homóloga34,35. Para investigar los efectos de la inhibición de MCP-1 in vivo en la AS precoz, en el presente estudio se diseñó de forma preliminar el plásmido de expresión eucariótica MCP-1 pSilencer2.0-U6-MCP-1-ARNsi, basado en los principios de ARNi.
Los vectores de expresión de ARNsi MCP-1 se transfectaron en un modelo de conejo de AS carotídea a través de una infusión local intraluminal, demostrando la menor disminución de MCP-1 local en el grupo ARNi que en el grupo modelo y grupo plásmido blanco tanto a nivel de ARNm como de proteínas, lo que indica la capacidad del ARNi para inhibir efectivamente la expresión de los genes al nivel de su transcripción. La hiperplasia intimal apareció como respuesta al acúmulo de células espumosas debajo de la capa endotelial, y como parámetro de evaluación, se usó el cociente I:M. Los resultados muestran que, comparado con el grupo modelo, el cociente I:M disminuyó significativamente en el grupo ARNi, mientras que no se detectó una diferencia significativa entre el grupo plásmido blanco y el grupo modelo. Esto sugirió que la regulación a la baja de la expresión de MCP-1 local con ARNi, la disminución de la migración de los monocitos en las capas subendoteliales y la fagocitosis de los lípidos dieron lugar a la resolución de la hiperplasia intimal y retrasaron eficazmente la progresión de la AS.
En el presente estudio, se empleó la técnica de ARNi in vivo para investigar los efectos del ARNsi MCP-1 sobre la AS local y precoz, lo que proporciona nuevos indicios para la prevención y el tratamiento de la aterosclerosis y sus complicaciones.
La presente investigación se financió a través del Key Induction Project of Science and Technology, Provincia de Guangdong (2005B31201001) y el Doctorate Funding Program for Higher Education, Ministry of Education, China (20050558053).