




Los mecanismos que promueven la progresión de la arteriopatía periférica (AP) son poco conocidos; sin embargo, dicha progresión puede estar causada por una disfunción inflamatoria subyacente. Por esta razón, en el presente estudio se examinó el perfil de las concentraciones de interleucina (IL)-1b, IL-2, IL-4, IL-6, IL-8, IL-10, IL-13, anticuerpos anticardiolipina y anticuerpos anti-β2-glucoproteína 1, y se caracterizó in vitro la respuesta inflamatoria de los sujetos a estudio. Se clasificó clínicamente a los individuos de la serie de acuerdo con los criterios de la Organización Mundial de la Salud y el índice de presión tobillo-brazo en: isquemia crítica (n = 20), claudicación estable (n = 20) y casos control (n = 20). Los estudios in vitro comprendieron el cultivo de sangre completa en medio RPMI-1640 durante 24 h, con y sin 1 mg/ml de lipopolisacárido, y la obtención del perfil de producción de citocinas. Los niveles de autoanticuerpos se determinaron mediante el estudio por enzimoinmunoanálisis de adsorción, mientras que los perfiles de citocinas se determinaron mediante inmunoanálisis múltiplex. Las concentraciones séricas de IL-6, IL-10, IL-13 y de anticuerpos anti-β2-glucoproteína 1 fueron más altas en los pacientes con AP. En el caso de las concentraciones de IL-6 y anticuerpos anti-β2-glucoproteína 1 los valores reflejaron una severidad de la enfermedad creciente (p < 0,05). Los estudios in vitro revelaron que la capacidad secretora de IL-8 e IL-13 es mayor de forma significativa en los casos de AP después de 6 h. Sin embargo, cuando se estandarizaron frente al recuento de leucocitos de los pacientes, los perfiles de producción de citocinas no difirieron. La AP se caracteriza por una mayor carga inflamatoria con independencia del tipo de citocina Th1:Th2, la cual es más pronunciada con una severidad de la enfermedad creciente. Sin embargo, es probable que la hiperreactividad inflamatoria de la sangre completa cultivada de pacientes con AP esté relacionada en mayor medida con una leucocitosis asociada que con una disfunción inflamatoria.
La arteriopatía periférica (AP) afecta al 10% de los hombres > 65 años de edad, y aumenta hasta el 20% en pacientes de ambos sexos ≥ 75 años de edad1. La dinámica y los estimulantes del flujo relacionados con la progresión de la enfermedad podrían diferir de los de las circulaciones coronaria y cerebral2. Los factores como la edad, el sexo, el consumo de tabaco, la diabetes, la hipertensión arterial, la concentración sérica de lipoproteínas de baja densidad y un índice de presión tobillo/brazo (IPTB) bajo han sido asociados con su progresión de forma independiente3-6.
No obstante, dichos factores no explican la progresión benigna de la enfermedad en la mayoría de los pacientes y su naturaleza agresiva en unos pocos7. Recientemente, su progresión se ha asociado con un aumento de los valores plasmáticos de fibrinógeno, proteína C reactiva, homocisteína y lipoproteína(a)8,9. Tras identificar la inflamación como un mediador de aterosclerosis, se han destacado una serie de citocinas de los linfocitos T colaboradores (T helper, Th)1 y Th2 como marcadores pronósticos de la progresión de la enfermedad10. A pesar de estos progresos, no se han publicado hasta la fecha estudios que hayan investigado el papel de las múltiples citocinas en relación con la gravedad de la AP.
Hay pruebas que respaldan una conexión entre un aumento de la concentración de IL-6 y los niveles de anticuerpos reactivos frente a las células endoteliales11. Éstos se dirigen contra los antígenos presentes en las membranas de la célula endotelial y tienen implicaciones en la aterosclerosis acelerada de los procesos autoinmunes12,13. Los anticuerpos anticardiolipina y su principal subpoblación, los anti-β2-glucoproteína 1, son dos de estos autoanticuerpos relacionados con la gravedad de la AP11. No obstante, apenas se conoce la relación entre sus perfiles y los de las citocinas Th1 y Th2 en la AP.
En consecuencia, el presente estudio presentó tres objetivos principales: 1) determinar los niveles séricos de IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, e IL-13; 2) obtener el perfil de las concentraciones de anticuerpos anticardiolipina y anti-β2-glucoproteína 1; y 3) caracterizar la respuesta inflamatoria (en relación con la capacidad secretora de IL-6, IL-8, IL-10, e IL-13) en pacientes con arteriopatía periférica e individuos control mediante un modelo de estimulación de sangre completa in vitro.
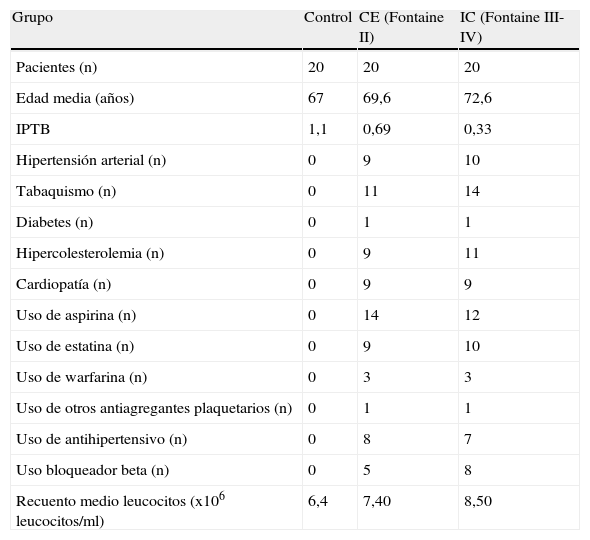
Materiales y MétodosPacientes del estudioLos pacientes fueron incluidos en el estudio tras la obtención de la aprobación de éste por un comité ético y la obtención de un consentimiento informado por escrito. Los subgrupos de AP fueron 1) pacientes con claudicación estable (CE, n = 20) con antecedentes de claudicación intermitente según la definición de la Organización Mundial de la Salud/cuestionario de Rose y un IPTB < 0,9 o, en el caso de los pacientes diabéticos, con una oclusión o una estenosis demostrada radio lógicamente de las arterias ilíaca, femoral o poplítea (angiografía o eco-Doppler) y 2) pacientes con isquemia crítica de la extremidad (IC, n = 20). La IC se definió como dolor en reposo isquémico crónico, úlceras o gangrena atribuibles a patología oclusiva arterial demostrada objetivamente (es decir, por una presión sistólica en el tobillo < 50 mmHg en pacientes con dolor en reposo o < 60 mmHg en úlceras de evolución tórpida y/o por una oclusión o una estenosis de la arteria ilíaca, femoral o poplítea demostradas radiológicamente). Los individuos control (n = 20) se incluyeron de forma sincrónica, emparejados por edad y sexo tras asegurar que no presentaban síntomas de AP y contaban con un IPTB de 0,9-1,2 o, en el caso de los sujetos diabéticos, con una presión en los dedos de los pies > 70 mmHg. Se excluyó a aquellos individuos que habían sufrido un infarto agudo de miocardio o un accidente vascular cerebral en los 3 meses previos, a los que participaban en otros estudios durante este período, o a los que tenían un diagnóstico confirmado de vasculitis, proceso autoinmune sistémico o cáncer. Las características demográficas de los pacientes estudiados se describen en la tabla I.
Demografía y características de los grupos a estudio
| Grupo | Control | CE (Fontaine II) | IC (Fontaine III-IV) |
| Pacientes (n) | 20 | 20 | 20 |
| Edad media (años) | 67 | 69,6 | 72,6 |
| IPTB | 1,1 | 0,69 | 0,33 |
| Hipertensión arterial (n) | 0 | 9 | 10 |
| Tabaquismo (n) | 0 | 11 | 14 |
| Diabetes (n) | 0 | 1 | 1 |
| Hipercolesterolemia (n) | 0 | 9 | 11 |
| Cardiopatía (n) | 0 | 9 | 9 |
| Uso de aspirina (n) | 0 | 14 | 12 |
| Uso de estatina (n) | 0 | 9 | 10 |
| Uso de warfarina (n) | 0 | 3 | 3 |
| Uso de otros antiagregantes plaquetarios (n) | 0 | 1 | 1 |
| Uso de antihipertensivo (n) | 0 | 8 | 7 |
| Uso bloqueador beta (n) | 0 | 5 | 8 |
| Recuento medio leucocitos (x106 leucocitos/ml) | 6,4 | 7,40 | 8,50 |
CE: claudicación estable; IC: isquemia crítica; IPTB: índice de presión tobillo/brazo.
Después de un ayuno nocturno de 12 h, los pacientes evitaron realizar ejercicio físico por la mañana y permanecieron en reposo durante 30 min antes de la obtención de una muestra de sangre completa mediante punción en la fosa antecubital con una aguja de calibre 16, que fue introducida en tubos heparinizados. La mayor parte se conservó para la estimulación con lipopolisacárido (LPS), mientras que se centrifugó 1 ml a 5.000 rpm durante 10 min para aislar el plasma. Éste se almacenó a –80 °C hasta el análisis para la determinación de los perfiles de citocinas circulantes.
Estimulación con LPS de la sangre completa in vitroLa estimulación con LPS es un análisis estándar para monitorizar la respuesta inflamatoria en una diversidad de células. Se diluyó la sangre completa heparinizada (4 ml) 1:1 con un medio RPM-1640 (con l-glutamina, sin rojo fenol; Invitrogen, Paisley, Reino Unido), suplementado con 2,5 U/ml de penicilina, 2,5 μl/ml de estreptomicina y 6,25 ng/ml de anfotericina B (Invitrogen), con (provocación con LPS) o sin (basal) 1 μg/ml de LPS (de Salmonella enterica serotipo Abortus equi) (Sigma-Aldrich, Poole, Dorset, Reino Unido) en placas de cultivo con ventilación (SLS, Nottingham, Reino Unido). Las placas se incubaron en un balancín de plataforma (SLS) para garantizar su mezcla a 37°C durante 24 h bajo una atmósfera humidificada de un 5% de CO2. Se obtuvieron muestras seriadas de 350 μl a las 0, 3, 6 y 24 h del cultivo y se centrifugaron a 5.000 rpm durante 3 min. El sobrenadante se congeló a –80°C hasta que se analizó para obtener el perfil de citocinas.
Perfil de citocinasLas concentraciones de citocinas se determinaron mediante inmunoanálisis multiplex de fase líquida usando equipos a medida (Upstate, Milton Keynes, Reino Unido), introducidos en un citómetro 100 Luminex (Austin, TX), provisto de un programa StarStation (versión 2.0; Applied Cytometry Systems, Dinnington, Reino Unido). Las muestras de plasma se analizaron para la interleucina (IL)-1β, IL-2, IL-4, IL-6, IL-8, IL-10 e IL-13, mientras que el cultivo de sangre completa se analizó para los niveles de IL-6, IL-8, IL-10 e IL-13.
Determinación de las concentraciones de anticuerpos anticardiolipina y anti-β2-glucoproteína 1Los anticuerpos anticardiolipina y anti-β2-glucoproteína 1 se determinaron mediante un sistema de enzimoinmunoanálisis de adsorción comercial (ELISA; Bindazyme, Birmingham, Reino Unido).
Presentación de los datos y análisis estadísticoLas concentraciones de citocinas se expresaron en picogramos por mililitro mientras que las de autoanticuerpos se proporcionaron en unidades por mililitro. La producción de citocinas in vitro se expresó como capacidad secretora (= perfil estimulado por LPS – niveles de producción basal [pg/ml]). Los valores también se estandarizaron de acuerdo con el recuento de leucocitos (picogramos/millón de leucocitos) puesto que se detectó un grado muy significativo de leucocitosis asociada a pacientes del subgrupo IC comparado con individuos de control (p < 0,001). Todos los datos se expresaron como media ± error estándar de la media. La significación entre grupos se evaluó con pruebas de Kruskall-Wallis y pruebas post hoc de Mann-Whitney, determinando la distribución no normal de los datos con pruebas de Anderson-Darling (Minitab, State Collage, PA, versión 14.1).
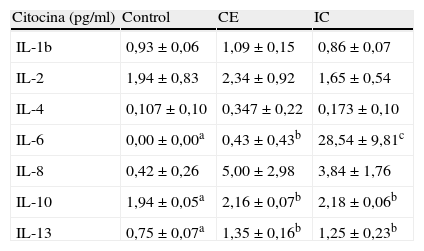
ResultadosPerfiles de citocinas circulantesSe observó un aumento marcado (66 veces) de los niveles de IL-6 (p < 0,001) en el grupo de pacientes con IC comparado con el grupo de pacientes con CE, en el que, a su vez, fue moderadamente más alto que en los casos control (p < 0,05), en los que prácticamente no se detectaron concentraciones de IL-6 (tabla II). Además, aunque las concentraciones circulantes de IL-10 e IL-13 fueron más altas (del orden de 1,1 y 2 veces, respectivamente) en ambos grupos de pacientes con AP comparado con los casos control (p < 0,05), no se detectaron diferencias evidentes en los niveles entre las poblaciones de pacientes con IC y CE. En comparación, en ninguna de las poblaciones del estudio hubo diferencias significativas en los perfiles de IL-1β, IL-2 e IL-8 circulantes.
Perfiles de citocinas séricas
| Citocina (pg/ml) | Control | CE | IC |
| IL-1b | 0,93 ± 0,06 | 1,09 ± 0,15 | 0,86 ± 0,07 |
| IL-2 | 1,94 ± 0,83 | 2,34 ± 0,92 | 1,65 ± 0,54 |
| IL-4 | 0,107 ± 0,10 | 0,347 ± 0,22 | 0,173 ± 0,10 |
| IL-6 | 0,00 ± 0,00a | 0,43 ± 0,43b | 28,54 ± 9,81c |
| IL-8 | 0,42 ± 0,26 | 5,00 ± 2,98 | 3,84 ± 1,76 |
| IL-10 | 1,94 ± 0,05a | 2,16 ± 0,07b | 2,18 ± 0,06b |
| IL-13 | 0,75 ± 0,07a | 1,35 ± 0,16b | 1,25 ± 0,23b |
Diferentes superíndices (a y b) indican diferencias estadísticamente significativas entre grupos, cp < 0,05.
CE: claudicación estable; IC: isquemia crítica.
Se observó un aumento de los anticuerpos anti-β2 glucoproteína 1 en asociación con una mayor gravedad de la AP (fig. 1). En particular, en los pacientes con IC los niveles fueron significativamente más altos que en los casos control (p < 0,05). Los niveles de anticuerpos anti-β2 glucoproteína 1 en los pacientes con CE fueron intermedios entre los del grupo de IC y el control y significativamente diferentes de estos dos grupos (p < 0,05). No obstante, la AP no afectó a los niveles de anticuerpos anticardiolipina en ninguna de las dos poblaciones (fig. 2).
. CI: isquemia crítica; Control: control; SC: claudicación estable.")

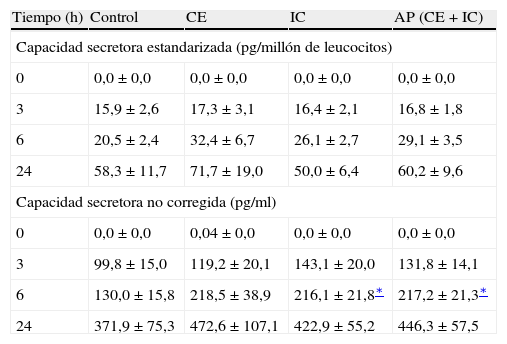
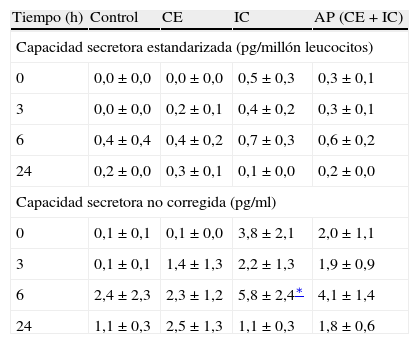
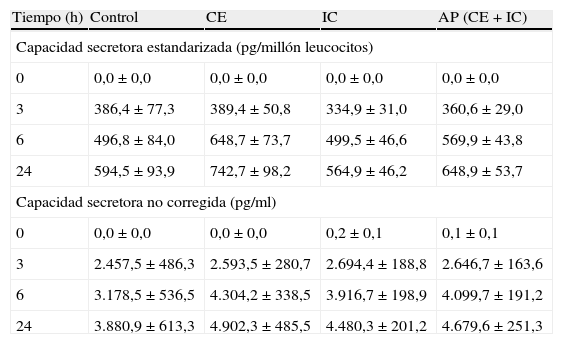
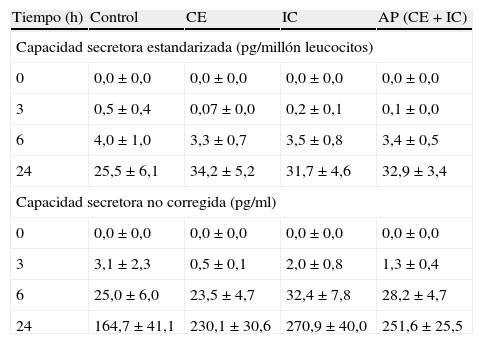
Tras estimulación con LPS in vitro, la capacidad secretora de IL-8 después de 6 h de incubación fue significativamente mayor en pacientes con IC al igual que en el conjunto de población con AP (es decir, CE + IC) (p < 0,05) (tabla III). Tras la estimulación con LPS in vitro, se observó una tendencia próxima a la significación estadística hacia una producción de IL-13 más alta en todos los pacientes con AP después de 6 h de incubación (p = 0,06). Sin embargo, sólo en la población IC se identificó una capacidad secretora de IL-13 significativamente mayor en este momento del estudio (p < 0,05) (tabla IV). En comparación, no se observó un aumento de las capacidades secretoras de IL-6 o IL-10 asociado a ninguno de los subgrupos de AP (tablas V y VI). Sin embargo, cuando se estandarizaron todas las capacidades secretoras para el recuento de leucocitos, no se identificó una diferencia significativa en el perfil de producción de citocinas entre los grupos de estudio.
Capacidades secretoras de IL-8 en los diferentes grupos a estudio
| Tiempo (h) | Control | CE | IC | AP (CE + IC) |
| Capacidad secretora estandarizada (pg/millón de leucocitos) | ||||
| 0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 |
| 3 | 15,9 ± 2,6 | 17,3 ± 3,1 | 16,4 ± 2,1 | 16,8 ± 1,8 |
| 6 | 20,5 ± 2,4 | 32,4 ± 6,7 | 26,1 ± 2,7 | 29,1 ± 3,5 |
| 24 | 58,3 ± 11,7 | 71,7 ± 19,0 | 50,0 ± 6,4 | 60,2 ± 9,6 |
| Capacidad secretora no corregida (pg/ml) | ||||
| 0 | 0,0 ± 0,0 | 0,04 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 |
| 3 | 99,8 ± 15,0 | 119,2 ± 20,1 | 143,1 ± 20,0 | 131,8 ± 14,1 |
| 6 | 130,0 ± 15,8 | 218,5 ± 38,9 | 216,1 ± 21,8∗ | 217,2 ± 21,3∗ |
| 24 | 371,9 ± 75,3 | 472,6 ± 107,1 | 422,9 ± 55,2 | 446,3 ± 57,5 |
AP: arteriopatía periférica; CE: claudicación estable; IC: isquemia crítica.
Capacidades secretoras de IL-13 en los diferentes grupos a estudio
| Tiempo (h) | Control | CE | IC | AP (CE + IC) |
| Capacidad secretora estandarizada (pg/millón leucocitos) | ||||
| 0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,5 ± 0,3 | 0,3 ± 0,1 |
| 3 | 0,0 ± 0,0 | 0,2 ± 0,1 | 0,4 ± 0,2 | 0,3 ± 0,1 |
| 6 | 0,4 ± 0,4 | 0,4 ± 0,2 | 0,7 ± 0,3 | 0,6 ± 0,2 |
| 24 | 0,2 ± 0,0 | 0,3 ± 0,1 | 0,1 ± 0,0 | 0,2 ± 0,0 |
| Capacidad secretora no corregida (pg/ml) | ||||
| 0 | 0,1 ± 0,1 | 0,1 ± 0,0 | 3,8 ± 2,1 | 2,0 ± 1,1 |
| 3 | 0,1 ± 0,1 | 1,4 ± 1,3 | 2,2 ± 1,3 | 1,9 ± 0,9 |
| 6 | 2,4 ± 2,3 | 2,3 ± 1,2 | 5,8 ± 2,4∗ | 4,1 ± 1,4 |
| 24 | 1,1 ± 0,3 | 2,5 ± 1,3 | 1,1 ± 0,3 | 1,8 ± 0,6 |
AP: arteriopatía periférica; CE: claudicación estable; IC: isquemia crítica.
Capacidades secretoras de IL-6 en los diferentes grupos a estudio
| Tiempo (h) | Control | CE | IC | AP (CE + IC) |
| Capacidad secretora estandarizada (pg/millón leucocitos) | ||||
| 0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 |
| 3 | 386,4 ± 77,3 | 389,4 ± 50,8 | 334,9 ± 31,0 | 360,6 ± 29,0 |
| 6 | 496,8 ± 84,0 | 648,7 ± 73,7 | 499,5 ± 46,6 | 569,9 ± 43,8 |
| 24 | 594,5 ± 93,9 | 742,7 ± 98,2 | 564,9 ± 46,2 | 648,9 ± 53,7 |
| Capacidad secretora no corregida (pg/ml) | ||||
| 0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,2 ± 0,1 | 0,1 ± 0,1 |
| 3 | 2.457,5 ± 486,3 | 2.593,5 ± 280,7 | 2.694,4 ± 188,8 | 2.646,7 ± 163,6 |
| 6 | 3.178,5 ± 536,5 | 4.304,2 ± 338,5 | 3.916,7 ± 198,9 | 4.099,7 ± 191,2 |
| 24 | 3.880,9 ± 613,3 | 4.902,3 ± 485,5 | 4.480,3 ± 201,2 | 4.679,6 ± 251,3 |
AP: arteriopatía periférica; CE: claudicación estable; IC: isquemia crítica.
Capacidades secretoras de IL-10 en los diferentes grupos a estudio
| Tiempo (h) | Control | CE | IC | AP (CE + IC) |
| Capacidad secretora estandarizada (pg/millón leucocitos) | ||||
| 0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 |
| 3 | 0,5 ± 0,4 | 0,07 ± 0,0 | 0,2 ± 0,1 | 0,1 ± 0,0 |
| 6 | 4,0 ± 1,0 | 3,3 ± 0,7 | 3,5 ± 0,8 | 3,4 ± 0,5 |
| 24 | 25,5 ± 6,1 | 34,2 ± 5,2 | 31,7 ± 4,6 | 32,9 ± 3,4 |
| Capacidad secretora no corregida (pg/ml) | ||||
| 0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 | 0,0 ± 0,0 |
| 3 | 3,1 ± 2,3 | 0,5 ± 0,1 | 2,0 ± 0,8 | 1,3 ± 0,4 |
| 6 | 25,0 ± 6,0 | 23,5 ± 4,7 | 32,4 ± 7,8 | 28,2 ± 4,7 |
| 24 | 164,7 ± 41,1 | 230,1 ± 30,6 | 270,9 ± 40,0 | 251,6 ± 25,5 |
AP: arteriopatía periférica; CE: claudicación estable; IC: isquemia crítica.
El aumento progresivo de la concentración circulante de IL-6 asociado a una severidad creciente de la AP coincide con lo descrito en estudios previos11,14. La IL-6 induce proteínas de fase aguda, como la proteína C reactiva (PCR), lo que explica sus mayores niveles circulantes y los de otras interleucinas, activa las plaquetas y aumenta la actividad procoagulante15,16. Aunque los niveles basales de IL-6 asociados a la isquemia crítica eran mayores, esto no se tradujo en una mayor capacidad secretora tras estimulación con LPS. Esto podría ser debido al hecho de que los mayores niveles circulantes reflejan un aumento de los niveles locales en los lugares de aterosclerosis17. Por otra parte, el aumento de los niveles plasmáticos de IL-6 podría ser atribuido a un origen celular diferente, como la lesión de las células endoteliales como consecuencia de la estimulación por anticuerpos antiendoteliales18. Este aumento de los niveles de IL-6 se pierde en el sistema in vitro, en parte a través de la dilución adicional de las muestras sanguíneas con RPMI y en parte a través de la estandarización de la capacidad secretora frente al perfil de leucocitos. La imposibilidad posterior del LPS para estimular una sobreproducción de IL-6 se opone al concepto de una respuesta inflamatoria sistémica aberrante que sería la base de la vulnerabilidad individual a la AP. No obstante, puesto que el modelo del presente estudio sólo presta atención a la sangre completa, es posible que cualquier supuesta disfunción inflamatoria subyacente pueda ser secundaria a un aumento de la producción de citocinas por los monocitos y/o a la activación de las células endoteliales19.
En la claudicación estable, los episodios inflamatorios repetidos que se producen asociados a la deambulación podrían contribuir a una inflamación crónica de bajo grado que diera lugar a la disfunción endotelial. Esto está respaldado por los estudios que demuestran una activación de los neutrófilos, un aumento en las moléculas de adhesión y la liberación de IL-8 después del ejercicio20-22. El aumento de los marcadores inflamatorios como la IL-6 podría reflejar así mismo una activación concomitante de bajo grado del sistema inmunitario23. En este contexto, un perfil inflamatorio más pronunciado podría causar una mayor carga aterosclerótica. Los niveles circulantes de IL-8 no estuvieron asociados a una mayor severidad de la AP, en comparación con lo descrito en publicaciones previas22, aunque estas discrepancias podrían ser explicadas por el hecho de que los trabajos citados estaban dirigidos al estudio de pacientes durante el ejercicio, lo que podría traducirse en la activación de los neutrófilos y en un aumento de la producción de citocinas tras el fenómeno de isquemia-reperfusión. Además, en el presente estudio la mitad de los pacientes (tabla I) recibía tratamiento con estatinas, para las cuales se ha identificado cada vez con mayor evidencia un efecto modulador de los niveles de marcadores inflamatorios, como son las citocinas24.
Después de la provocación con LPS, la capacidad secretora de IL-8 aumentó tras 6 h de incubación en la población AP en general y en los pacientes con IC en particular. Si este aumento precoz de la producción de IL-8 también tiene lugar después de la estimulación inflamatoria in vivo, podría contribuir al quimiotactismo de los neutrófilos hacia áreas inflamatorias, como las placas ateroscleróticas, donde también podría desempeñar un papel al desencadenar una adherencia firme de los monocitos en el endotelio vascular durante la etapa de rodamiento y durante su infiltración en la pared del vaso, como parte de los estadios precoces en la progresión de la aterosclerosis25.
En el presente estudio se demostró un aumento de los niveles circulantes de IL-10 en ambas poblaciones de AP en comparación con individuos de control, lo que contrasta con los hallazgos de DePalma et al26. Más que respaldar un desequilibrio Th1:Th2 como mecanismo subyacente causante de la AP, estos hallazgos respaldan un papel regulador de la IL-10, la cual compensa los mayores niveles de citocinas de tipo Th1 como la IL-6. La presente investigación también es el primer estudio que determina los niveles de IL-13 en la AP. La IL-13 estuvo presente en concentraciones bajas y en gran parte su producción no fue sensible a la provocación con LPS in vitro. A pesar de que se observó un aumento significativo de los niveles circulantes de IL-13 asociado a la AP, dicho aumento fue cuantitativamente menor. Esto podría reflejar simplemente un mecanismo modulador de bajo grado de tipo Th2, establecido para contrarrestar los mayores niveles de IL-6 asociados a AP, tal y como se ha descrito para la IL-10. Por otra parte, podría reflejar unas propiedades paradójicamente proinflamatorias de la IL-13, según lo demostrado por la potenciación de la expresión del receptor de IL-8, la oxidación de lipoproteínas de baja densidad (LDL) por parte de los monocitos y la potenciación de la transmigración de los leucocitos27-29.
Los bajos niveles circulantes de IL-1β e IL-2 observados en todos los grupos de estudio sugieren que estas citocinas podrían desempeñar un papel limitado en la fisiopatología de la AP. Aunque estos niveles bajos coinciden con los hallazgos previos26,30, también es posible que las concentraciones periféricas no sean representativas de su participación local en las placas ateroscleróticas. Los niveles circulantes de IL-4 antiaterogénica fueron de igual manera muy bajos en todos los grupos, quizá porque su expresión en la aterosclerosis es limitada31.
Existen diversos desencadenantes inflamatorios potenciales en la AP. Los pacientes con AP y una mayor carga aterosclerótica presentan probablemente mayores niveles de anticuerpos frente a las LDL oxidadas32. Estos mismos anticuerpos podrían reaccionar de forma cruzada contra las células endoteliales junto con los anticuerpos anticardiolipina, β2-glucoproteína 1, y las proteínas del choque térmico33. Aunque se ha demostrado que unos niveles elevados de anticuerpos anticardiolipina son un factor de riesgo independiente de infarto de miocardio y mortalidad cardíaca en hombres de mediana edad34, no se observó un aumento de dichos anticuerpos en los pacientes con AP del presente estudio. Otros estudios publicados respaldan estos hallazgos negativos35,36. En el presente estudio, se observó un aumento de los antiβ2 glucoproteína 1 en pacientes con IC y CE comparado con individuos control. Estos datos coinciden con los estudios que demuestran un aumento de los niveles de este anticuerpo, en diversos estudios en animales y seres humanos que revelan la correlación entre el aumento de los títulos de anticuerpos antiβ2 glucoproteína 1 y una aterosclerosis acelerada11,13,37.
Otra fuente potencial de inflamación en la AP podría guardar relación con el ejercicio. En pacientes con claudicación, los episodios inflamatorios repetidos asociados a la deambulación podrían dar lugar a un proceso inflamatorio de bajo grado y a disfunción endotelial. Esta propuesta está respaldada por estudios que señalan una activación de neutrófilos, al igual que un aumento en la producción de moléculas de adhesión e IL-8 después del ejercicio21,22,38. Por último, la inflamación también podría reflejar una activación del sistema inmunitario de bajo grado23. En este contexto, el perfil inflamatorio más pronunciado de los pacientes con claudicación podría ser tanto la causa como la consecuencia de una mayor carga aterosclerótica.
En conclusión, el presente estudio no respalda que las citocinas Th1 o Th2 sean predominantes en la arteriopatía periférica (es decir, representando un desequilibrio Th1:Th2), aunque la carga inflamatoria es más pronunciada en las manifestaciones graves de la enfermedad. El menor grado de hiperreactividad inflamatoria asociada con la sangre completa cultivada observado en pacientes con CE e IC pareció estar relacionado con la presencia de leucocitosis más que por la existencia de una disfunción inflamatoria inherente per se. El aumento de los niveles de anticuerpos reactivos contra la célula endotelial en la AP junto con sus acciones estimulando el incremento de los niveles de IL-6 podrían contribuir a la presencia de una mayor carga inflamatoria y ser responsables de la aterosclerosis acelerada observada en pacientes con isquemia critica.