





La microangiopatía trombótica (MAT) es un síndrome que se caracteriza por la presencia de anemia hemolítica microangiopática, trombocitopenia y disfunción orgánica de severidad variable que puede ser consecuencia de una constelación heterogénea de causas. Estudios preliminares han mostrado altas tasas de secuelas a largo plazo, como la enfermedad renal crónica y una mortalidad que varía entre el 30 y el 90% dependiendo del momento del diagnóstico y de la terapia utilizada.
MétodosSe incluyeron 6 pacientes que ingresaron a las unidades de cuidado intensivo del Hospital Santa Clara de enero a diciembre de 2017 con diagnóstico de MAT definida como la presencia de trombocitopenia (<150.000 plaquetas/mm3) o disminución mayor al 25% del recuento inicial, anemia hemolítica microangiopática y disfunción orgánica. Se analizaron variables demográficas, abordaje diagnóstico y tratamiento, así como variables asociadas con mortalidad.
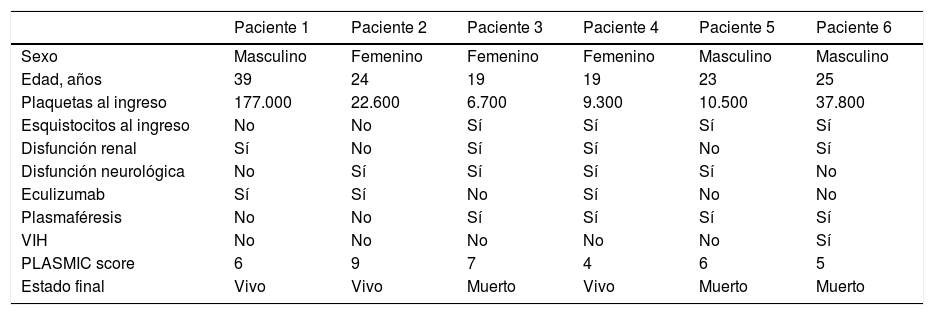
ResultadosSe describe el comportamiento clínico de 6 pacientes. Edad de 25±7,2años, hombres 50%. El 66% de los pacientes (n=4) tuvieron presencia de esquistocitos en el extendido de sangre periférica desde el ingreso, el 34% restante (n=2) presentaron esquistocitos en las siguientes 24h. El 66% de los pacientes (n=4) presentaron disfunción renal y neurológica, el 83% (n=5) presentaron trombocitopenia al ingreso, el 66% recibieron terapia de recambio plasmático, con mortalidad en el 50% de estos casos. El 83% de los pacientes requirieron ventilación mecánica invasiva. En un paciente no fue posible la toma de ADAMTS13, por haber recibido transfusión de plaquetas ante alto riesgo de sangrado. Los 3 pacientes sobrevivientes recibieron tratamiento con eculizumab.
ConclusionesLa MAT es una entidad que atenta contra la vida; debe ser sospechada en todo paciente que se encuentra con disfunción orgánica y trombocitopenia y hacerse la búsqueda activa de la misma mediante el conjunto de hallazgos clínicos más comunes (alteraciones neurológicas y renales, entre muchos otros) y paraclínicos (esquistocitos). Los buenos resultados y la sobrevida dependen del inicio temprano del tratamiento. Esta entidad es poco sospechada, diagnosticada y tratada tempranamente en las unidades de cuidado intensivo.
TMA is a syndrome characterized by microangiopathic hemolytic anemia, thrombocytopenia and multiple organ failure with variable severity as consequence of a constellation of heterogeneous causes. Preliminary information shows a high rate of long term sequelae as chronic kidney disease and mortality rate that varies between 30% and 90% depending on the time of diagnosis and therapy used.
MethodsWe described six patients admitted to the intensive care unit of Santa Clara Hospital from Januaray to december of 2017 with a diagnosis of TMA defined as the presence of thrombocytopenia (<150.000 platelets per ml3) or a decrease grater than 25% of the initial count, microangiopathyc hemolytic anemia and multiple organ dysfunction. Demographic variables and diagnostic approach were analyzed as well as variables associated with mortality.
ResultsThe clinical behavior of 6 patients was described. Age of 25±7.2 years, men 50%. 66% of the patients had schistocytes in the peripheral smear from admission, the remaining 34% presented schistocytes in the next 24 hours. 66% of the patients present with kidney and neurological dysfunction, 83% presented thrombocytopenia at admission, 66% received plasma exchange therapy with mortality in 50% of these cases. 83% of patients required invasive mechanical ventilation. In a patient it was not possible measure ADAMTS13 activity due to platelet transfusion. The 3 surviving patients received treatment with eculizumab.
ConclusionTMA is an entity that threats life, should be suspected in every patient with organic dysfunction and thrombocytopenia in the intensive care unit and make the active search for it through more common clinical findings (neurological and kidney dsyfunction among many others) and paraclinics (schistocytes), survival depends on the early onset of treatment. This entity is not commonly suspected, diagnosed and treated early in the intensive care units.
Artículo
Socios de la Asociación de Medicina Crítica y Cuidado Intensivo
![]()
Para acceder a la revista
Es necesario que lo haga desde la zona privada de la web de la AMCI, clique aquí
