






Amyotrophic lateral sclerosis (ALS) is the most common neurodegenerative disease affecting motor neurons. Although a small proportion of ALS cases are familial in origin and linked to mutations in specific genes, most cases are sporadic and have a multifactorial aetiology. Some recent studies have increased our knowledge of ALS pathogenesis and raised the question of whether this disorder is a proteinopathy, a ribonucleopathy, an axonopathy, or a disease related to the neuronal microenvironment.
DevelopmentThis article presents a review of ALS pathogenesis. To this end, we have reviewed published articles describing either ALS patients or ALS animal models and we discuss how the main cellular pathways (gene processing, protein metabolism, oxidative stress, axonal transport, relationship with neuronal microenvironment) may be involved in motor neurons degeneration.
ConclusionsALS pathogenesis has not been fully elucidated. Recent studies suggest that although initial triggers may differ among patients, the final motor neurons degeneration mechanisms are similar in most patients once the disease is fully established.
La esclerosis lateral amiotrófica (ELA) es la enfermedad degenerativa de las motoneuronas más frecuente. Aunque un pequeño porcentaje de los casos de ELA tienen un origen familiar y son secundarios a mutaciones en genes concretos, a la gran mayoría de ellos se les presupone un origen multifactorial, sin que su patogenia haya sido completamente aclarada. No obstante, en los últimos años varios estudios han aumentado el conocimiento sobre la patogenia de la enfermedad, planteando la cuestión de si se trata de una proteinopatía, una ribonucleinopatía, una axonopatía o una enfermedad del microambiente neuronal.
DesarrolloEn el presente artículo revisamos los trabajos publicados tanto en pacientes como en modelos animales de ELA y discutimos la implicación de los principales procesos celulares que parecen contribuir a su patogenia (procesamiento génico, metabolismo de proteínas, estrés oxidativo, transporte axonal y relación con el microambiente neuronal).
ConclusionesAunque la patogenia de la ELA dista de estar aclarada, los estudios recientes apuntan a la idea de que hay unos desencadenantes iniciales que varían de unos sujetos a otros, y unas vías finales de degeneración de las motoneuronas que están implicadas en la mayor parte de los casos de enfermedad.
Amyotrophic lateral sclerosis (ALS) is the most frequent degenerative motor neuron disease, with incidence ranging from 1 to 3 cases per 100000 person-years.1,2 Although cases have been reported in patients in the second and third decades of life, peak incidence occurs between the ages of 60 and 70.3 ALS is characterised by involvement of both the upper and the lower motor neurons, which causes the typical symptoms of the disease: muscle weakness, atrophy, fasciculations, and spasticity. There are 2 types of ALS: familial ALS (fALS), which represents 5% to 10% of cases, is characterised by familial aggregation and normally follows an autosomal dominant inheritance pattern; and sporadic ALS (sALS), which is not clearly associated with a family history of the disease and is therefore believed to be sporadic in origin.3 Familial ALS is secondary to mutations in genes directly associated with motor neuron degeneration, whereas sporadic forms are though to be caused by multiple factors. This study aims to review the different dysfunctional cellular pathways in ALS and to analyse how such alterations predispose to motor neuron degeneration.
DevelopmentALS specifically affects cells, leading to tissue alterations; the most frequently affected cells are motor neurons (Fig. 1). Although the precise factors determining preferential involvement of motor neurons are yet to be fully understood, certain factors have been associated with the particular vulnerability of these cells: (1) the large size of the cells and their robust cytoskeleton, which has high metabolic demands to maintain cell functions; (2) high reliance on optimal mitochondrial function; (3) high vulnerability to excitotoxicity and dysregulation of intracellular calcium homeostasis; and (4) reduced capacity for heat shock response and chaperone activity, and possibly reduced function of the ubiquitin proteasome system.4 Although ALS involves multiple pathogenic mechanisms which are yet to be determined, a number of genetic factors and alterations in the main cellular pathways have been characterised and associated with disease onset. These factors will be addressed in the following sections (Fig. 2).

 Alterations at the level of RNA processing, resulting in aberrant and/or toxic RNA. (2) High levels of oxidative stress and difficulty eliminating free radicals. (3) Protein metabolism alterations associated with UPS inhibition/dysfunction and hyperactivation of autophagy. (4) Alterations in axonal transport proteins. (5) Alterations in glial cells leading to motor neuron degeneration.")
Pathogenic alterations in ALS. The diagram displays the multiple cell function alterations that have been described in patients with ALS. (1) Alterations at the level of RNA processing, resulting in aberrant and/or toxic RNA. (2) High levels of oxidative stress and difficulty eliminating free radicals. (3) Protein metabolism alterations associated with UPS inhibition/dysfunction and hyperactivation of autophagy. (4) Alterations in axonal transport proteins. (5) Alterations in glial cells leading to motor neuron degeneration.
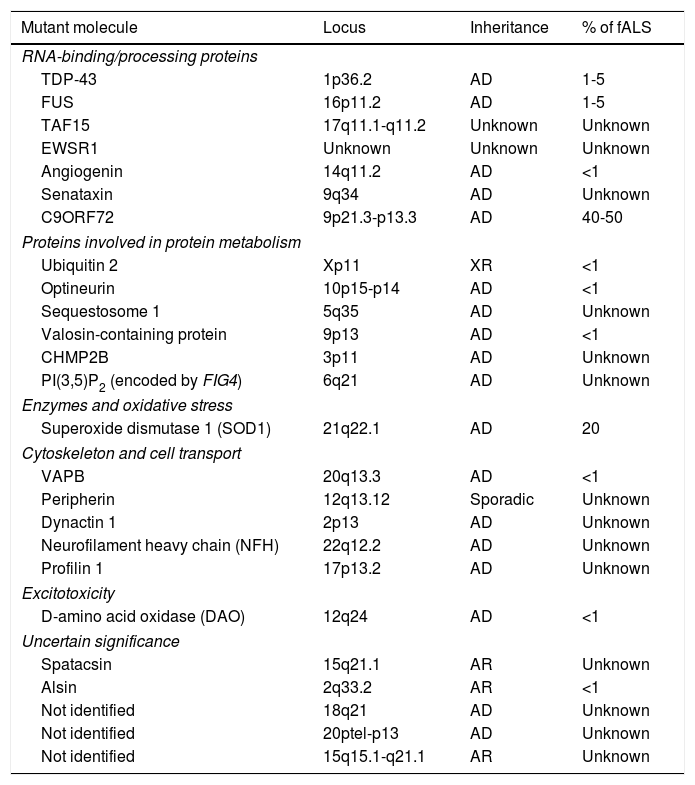
Familial forms of ALS account for 5% to 10% of cases and are frequently associated with an autosomal dominant inheritance pattern. The most frequent forms of fALS are secondary to mutations in the superoxide dismutase 1 (SOD1) gene and mutations on chromosome 9. Table 1 lists the mutations identified to date in patients with fALS. The study of familial cases of ALS is essential since it opens the door to new hypotheses on the aetiopathogenesis of the disease which may be extrapolated to sALS.4
Genes involved in the pathogenesis of some forms of familial ALS.
| Mutant molecule | Locus | Inheritance | % of fALS |
|---|---|---|---|
| RNA-binding/processing proteins | |||
| TDP-43 | 1p36.2 | AD | 1-5 |
| FUS | 16p11.2 | AD | 1-5 |
| TAF15 | 17q11.1-q11.2 | Unknown | Unknown |
| EWSR1 | Unknown | Unknown | Unknown |
| Angiogenin | 14q11.2 | AD | <1 |
| Senataxin | 9q34 | AD | Unknown |
| C9ORF72 | 9p21.3-p13.3 | AD | 40-50 |
| Proteins involved in protein metabolism | |||
| Ubiquitin 2 | Xp11 | XR | <1 |
| Optineurin | 10p15-p14 | AD | <1 |
| Sequestosome 1 | 5q35 | AD | Unknown |
| Valosin-containing protein | 9p13 | AD | <1 |
| CHMP2B | 3p11 | AD | Unknown |
| PI(3,5)P2 (encoded by FIG4) | 6q21 | AD | Unknown |
| Enzymes and oxidative stress | |||
| Superoxide dismutase 1 (SOD1) | 21q22.1 | AD | 20 |
| Cytoskeleton and cell transport | |||
| VAPB | 20q13.3 | AD | <1 |
| Peripherin | 12q13.12 | Sporadic | Unknown |
| Dynactin 1 | 2p13 | AD | Unknown |
| Neurofilament heavy chain (NFH) | 22q12.2 | AD | Unknown |
| Profilin 1 | 17p13.2 | AD | Unknown |
| Excitotoxicity | |||
| D-amino acid oxidase (DAO) | 12q24 | AD | <1 |
| Uncertain significance | |||
| Spatacsin | 15q21.1 | AR | Unknown |
| Alsin | 2q33.2 | AR | <1 |
| Not identified | 18q21 | AD | Unknown |
| Not identified | 20ptel-p13 | AD | Unknown |
| Not identified | 15q15.1-q21.1 | AR | Unknown |
Some mutations affect the genes coding for the vascular endothelial growth factor and the hereditary haemochromatosis protein. Copy number variants have also been described in the survival motor neuron genes (SMN1 and SMN2).5 However, the correlation of these mutations with ALS has not been fully replicated in subsequent studies of other populations. The study of candidate genes in patients with sporadic forms of ALS has identified several polymorphisms associated with increased risk of the disease. Several genome-wide association studies of ALS have been published to date. These studies have identified several loci associated with increased susceptibility to the disease, including those containing the FGGY carbohydrate kinase domain containing gene (FGGY), the dipeptidyl peptidase like 6 gene (DPP6), or the 1,4,5-triphosphate receptor type 2 gene (ITPR2).6–8 However, most of these findings have not been replicated in subsequent studies with larger samples. The pathogenic role of other genes, such as the gene coding for protein unc-13 homologue A (UNC13A), seems more robust, although this association is yet to be replicated in other studies.9 In 2010, an intronic expansion in C9ORF72 was identified in a high percentage of patients with both sporadic and familiar forms of ALS; until then, this was classified as chromosome 9p-linked ALS.10,11 This expansion has also been described in patients with frontotemporal dementia (FTD) and those with ALS-FTD.12,13 This highly relevant finding is addressed in detail in a later section.
Until now, genetic studies have mainly focused on European populations. Future genetic studies including patients of different ethnic origins and analyses of rare variants (that is, polymorphisms with an allele frequency<5%) will enable the identification of new pathogenic loci. A recent exome sequencing study has revealed de novo mutations in some genes mainly involved in forms of fALS, such as those secondary to mutations in SOD and in the fused-in sarcoma gene (FUS). However, not all of these are necessarily pathogenic.14–16 The field of exome sequencing promises to provide valuable new information in the coming years.
Transcriptional alterations and alterations in RNA processingA large number of proteins which have been linked to ALS are directly or indirectly involved in nucleic acid processing. The first findings came after mutations in SMN1 were identified as being responsible for spinal muscular atrophy.17 The SMN protein is vital for the survival of motor neurons. This protein is involved in pre-mRNA splicing18 and the axonal transport of mRNA for subsequent translation in the presynaptic terminal, more specifically in the neuromuscular junction.18,19
Subsequent identification of mutations in TDP-43 and FUS confirmed that aberrant RNA metabolism may contribute to the pathogenesis of ALS.
TAR-DNA binding protein 43Mutations in the TARDBP gene, which codes for the TAR-DNA binding protein 43 (TDP-43), are a rare cause of fALS, following an autosomal dominant inheritance pattern.20 Research into TDP-43 as a candidate gene began after intracellular TDP-43 aggregates were found in a majority of patients with both sporadic and familial ALS.21 Strikingly, these aggregates are not seen in cases secondary to SOD or FUS mutations.21 TDP-43 is a protein of the ribonucleoprotein family, present predominantly in the cell nucleus. It interacts with DNA and RNA molecules, regulating the transcription, splicing, and subsequent transport of RNA.22 Most mutations of the TARDBP gene are located in exon 6 and constitute nonsense mutations.23 The TARDBP protein has 2 prion-like domains which bind to GU-rich intronic regions in RNA molecules, interacting with over 6000 different RNA messengers.22 During situations of stress, TDP-43 remains mainly in the cytoplasm, forming cytoplasmatic aggregates known as stress granules.24 To prioritise the translation of proteins that are essential to cell survival, these aggregates sequester RNA molecules that are not involved in the neuronal stress response, blocking their translation until the stressor is no longer present. Aggregates then dissociate due to the action of peptidases and chaperones, and TDP-43 returns to the nuclear compartment.25 The mechanisms by which TDP-43 mutations lead to the development of ALS are yet to be determined. Mutations associated with the development of the disease increase the protein's translocation from the nucleus to the cytoplasm and promote aggregation. The abnormal protein will therefore be unable to play its normal role in RNA transcription and processing, promoting aggregation in the cytoplasm and the sequestration of other molecules necessary for cell homeostasis.22,26,27
FET protein family: the FUS proteinGenes coding for proteins of the FET family are also associated with gene processing and involved in the pathogenesis of ALS. The FET family includes the TAF15 protein (TATA box-binding protein-associated factor 15), EWSR1 (Ewing sarcoma breakpoint region 1), and FUS.28
FUS is a DNA- and RNA-binding nuclear protein involved in transcriptional regulation, DNA and RNA processing, and mRNA transport. FUS mutations account for 4% of all cases of fALS and fewer than 1% of cases of sALS.21 Most FUS mutations are located on exons 13 and 15, which code for a nuclear localisation signal. These mutations cause abnormal translocation to the cytoplasm, leading to the recruitment of FUS into stress granules.29,30 As in the case of TDP-43, FUS interacts with a wide range of genes, possibly over 5500, through a prion-like domain; it would therefore seem to affect a large number of cell processes.31 Although the exact mechanisms of FUS cytotoxicity are yet to be understood, it has been suggested that gain- and loss-of-function phenomena may coexist, as occurs with TDP-43.
Mutations in the C9ORF72 geneC9ORF72 contains the hexanucleotide repeat GGGGCC, which is located between 2 transcription start sites. The C9ORF72 hexanucleotide repeat expansion has been found in up to 40% of families with ALS and in 7% of patients with sALS.32 As previously mentioned, this expansion has also been reported in patients with FTD with and without ALS.10,32 Healthy individuals normally carry 2-5 repeats, and never over 30. The function of C9ORF72 is unknown. The protein is known to be structurally related to the family of DENN (differentially expressed in normal and neoplastic cells) domain proteins.33 From a functional viewpoint, the DENN domain is involved in the regulation of a series of GTPases, which use and degrade guanosine triphosphate (GTP).34 Penetrance of this mutation is estimated at 50% by the age of 60 and nearly 100% after the age of 80.32 The pathogenic mechanism by which an excessive number of repeats is associated with disease development is yet to be determined. Numerous questions remain to be answered, for example whether homozygosity or a larger number of repeats are associated with more aggressive phenotypes. Some studies including patients with this expansion have shown that their levels of C9ORF72 mRNA were up to 50% lower, which suggests that the mutant allele is probably unable to produce mature RNA; this would be linked to the hypothesis of a loss-of-function process.35 On the other hand, in situ hybridisation studies of ALS patients carrying this expansion have demonstrated the presence of nuclear RNA aggregates in the cortex and the spinal cord, which points to a toxic gain-of-function mechanism.10
Other proteins involved in gene processingThough less important, other proteins involved in gene processing, such as angiogenin and senataxin, have also been found to be altered in patients with ALS.36
Alterations in protein metabolismProtein synthesisThe nucleolus and the endoplasmic reticulum (ER), which synthesise ribosomal RNA and proteins, respectively, together with the ubiquitin proteasome system (UPS) and the autophagy-lysosome system, which play a major role in protein elimination, are the main elements involved in protein metabolism in human cells. Most neurodegenerative diseases are characterised by cellular accumulation of aberrant or misfolded proteins. In the case of ALS, it has been suggested that such aggregates and their oligomeric precursors alter normal cell function, inducing a high level of oxidative stress which interferes with the cell's basic function, ultimately leading to cell death.37
Some of the ER's major functions include protein synthesis, folding, and “quality control”. Cellular stress may induce an ER stress response, characterised by chromatolysis and accumulation of aberrant and misfolded proteins in cytoplasmic inclusions. Chromatolysis is the fragmentation and subsequent cytoplasmic dissolution of ER cisternae. A number of studies have reported chromatolysis and protein aggregates both in patients with ALS and in experimental models (Fig. 3).38–41 ER disaggregation results in alterations in this structure's ability to synthesise proteins, frequently leading to the activation of apoptotic mechanisms.39 Ribosomes constitute a fundamental part of the rough ER; ribosome biogenesis is therefore essential to maintain the structural and functional integrity of this organelle. The process of ribosome synthesis is strictly regulated and nucleolus-mediated.42–44 The nucleolus therefore plays a crucial role in coordinating ER stress responses.45–48 Disrupted nucleolar function has been associated with some neurodegenerative diseases.49–53 Some researchers have observed increased nucleolar diameter in a transgenic model of ALS, which may be linked to the cells’ attempt to increase ribosomal gene synthesis in response to stress.54
 and a transgenic mouse model of ALS (SOD1-G93A) (B). The motor neuron of the healthy mouse shows no cytoplasmic inclusions whereas that of the transgenic mouse displays multiple polyubiquitinated aggregates secondary to proteasomal degradation.")
Ubiquitin-stained motor neurons from a healthy control mouse (A) and a transgenic mouse model of ALS (SOD1-G93A) (B). The motor neuron of the healthy mouse shows no cytoplasmic inclusions whereas that of the transgenic mouse displays multiple polyubiquitinated aggregates secondary to proteasomal degradation.
New forms of fALS secondary to mutations in genes coding for proteins directly involved in proteostasis have been described in recent years. The UPS and the autophagy-lysosome system constitute the 2 major cellular proteolytic pathways. Due to functional overlap between these protein degradation pathways, it is difficult to determine which plays the primary role in cell dysfunction. Some experimental studies have attempted to use mouse models to evaluate the role of each of these pathways separately. These studies showed how proteasome inhibition was correlated with neuronal loss and the accumulation of cells rich in TDP-43, OPTN, and FUS; these effects were not observed after inhibiting autophagy.55 These findings suggest that motor neuron homeostasis in ALS may be more compromised in cases of UPS dysfunction than in those of autophagy dysfunction. However, this conclusion should be interpreted with caution and confirmed in future studies. In any case, there is solid evidence that proteostasis dysfunction secondary to UPS and autophagy malfunction contributes to the loss of neuronal homeostasis. Addressed below are particular mutations of genes involved in protein degradation pathways, identified in patients with ALS.
Ubiquilin-2Ubiquilin-2 (UBQLN2) is a ubiquitin-like protein involved in proteostasis which promotes proteasomal proteolysis.56 Mutations in the gene coding for this protein (UBQLN2) were found several years ago in patients with Alzheimer disease.57 Mutations in UBQLN2 have recently been found to play a role in the pathogenesis of some sporadic cases of ALS and ALS-FTD; UBQLN2 inclusions have been observed in motor neurons of the anterior horn in patients with ALS. These inclusions have also been observed in ALS patients with no mutations in UBQLN2, which suggests that UBQLN2 may play an important role in the degeneration mechanisms of motor neurons.58
Valosin-containing proteinExome sequencing studies have identified nonsense mutations in the gene coding for valosin-containing protein (VCP) in patients with fALS.59 VCP is a chaperone involved in multiple biological processes, including proteostasis.60 In this process, VCP participates in protein degradation at the level of the UPS and the autophagy-lysosome system.61
CHMP2B, optineurin, and PI(3,5)P2 5-phosphataseMutations in the genes coding for the charged multivesicular body protein 2B (CHMP2B), optineurin, and phosphatidylinositol 3,5-bisphosphate (PI[3,5]P2) 5-phosphatase have been found in some patients with fALS.62–64 CHMP2B is a protein involved in autophagic clearance.65 Optineurin, on the other hand, regulates multiple cell processes, including membrane trafficking and protein secretion.66 Lastly, mutations in factor-induced gene 4 (FIG4), which codes for PI(3,5)P2 5-phosphatase, have been reported in a small series of patients with ALS. This enzyme is located in the endosome membrane, where it mediates lysosomal function.67
SOD1 and oxidative stressSOD1 is a cytosolic enzyme containing 150 amino acids, a copper atom, and a zinc atom. It catalyses the conversion of superoxide into oxygen and hydrogen peroxide, which explains its importance in the antioxidant defence of cells with aerobic metabolism, as is the case of motor neurons. In fact, SOD1 mutations are present in 20% of patients with fALS. Over 150 different mutations have been described to date, of which most are nonsense mutations and nearly all follow an autosomal dominant inheritance pattern.23SOD1 mutations result in considerable SOD1 misfolding, which causes the protein to be processed for degradation by the UPS. However, a large proportion of misfolded proteins will not be degraded by the UPS, interfering with proteostasis68,69 and causing secondary autophagic activation. Both mouse models and patients with ALS have been shown to have increased numbers of autophagosomes.70,71 Progressive accumulation of the misfolded mutant protein, combined with the action of other stress factors such as ageing, induces a cell stress response.72 SOD1 misfolding is not exclusive to mutant forms of the protein. It has been demonstrated that oxidative stress leads to misfolding and aggregation of the wild-type SOD1 protein, as occurs with the mutant protein.73,74 These findings suggest that SOD1 may also play a role in sporadic forms of ALS, contributing to disease pathogenesis once cell stress is established by other mechanisms.
Oxidative stress occurs when there is an imbalance between free radical generation and elimination or when the cell is unable to repair or eliminate damage caused by this stress. Various studies suggest that oxidative stress is involved in ALS pathogenesis. Blood, urine, and cerebrospinal fluid (CSF) samples from patients with sALS have revealed increased levels of markers of free radical damage.75–77 Autopsy studies have also confirmed the presence of protein, lipid, and DNA alterations secondary to oxygen radicals in both sALS and in fALS secondary to SOD1 mutations.78–80 Experimental studies with mouse models of mutant SOD1 have shown increased levels of oxidation of different mRNA in presymptomatic stages of the disease, with a correlation between this oxidation and decreased expression of the protein. The most widely accepted hypothesis at present is that in addition to the damage it causes directly, oxidative stress promotes other pathogenic mechanisms which contribute to neuronal alterations, including excitotoxicity, protein aggregation, ER stress response, and mitochondrial dysfunction, and it also affects the interaction between motor neurons and the neuronal microenvironment.
Alterations in axonal transportGiven the length of motor neuron axons, axonal transport constitutes a key feature in the biology of these cells. Axonal transport, which is ATP-dependent, is essential to the supply of necessary cell components (RNA, proteins, and various different organelles) to the presynaptic terminal. The machinery of axonal transport comprises mainly microtubules and motor proteins, kinesins, and dyneins, which are associated to microtubules.72,81 Several studies in transgenic mice and patients with ALS have reported denervation and axon retraction in very early stages of the disease, even before neuronal loss becomes evident.82,83 Studies with a mouse model of mutant SOD1 have shown alterations in anterograde and retrograde transport in early stages of ALS and how these alterations are cargo-dependent; anterograde mitochondrial transport is especially compromised.83–85 Alterations in axonal transport have traditionally been thought to be secondary to multiple disorders, including mitochondrial dysfunction, resulting in insufficient ATP production, disrupted kinesin function, and protein aggregation.86–88 In recent years, however, alterations have also been identified in genes coding for proteins directly involved in axonal transport. Some patients with ALS have shown decreased neurofilament light chain expression.89 Strikingly, some researchers have reported that neurofilament light chain mRNA is preferentially sequestered within stress granules in the motor neurons of patients with ALS.90 Another recent finding is the description of mutations in the profilin 1 gene (PFN1).91 Profilin 1 is essential for actin filament polymerisation; PFN1 mutations therefore inhibit axonal growth, promoting axonal retraction and denervation. Some patients with ALS have also been found to have mutations in the genes coding for the neurofilament heavy chain, peripherin, dynactin, and alsin, among other proteins.92–95 In contrast, it has been suggested that those factors promoting axonal growth and regeneration, such as the vascular endothelial growth factor, may have a neuroprotective effect.96,97
Lastly, recent GWAS have demonstrated a link between some cases of sALS and particular polymorphisms in genes coding for such axonal proteins as kinesin-associated protein 3 and acetyltransferase complex subunits.98,99
ExcitotoxicityGlutamate is the principal excitatory neurotransmitter in the central nervous system (CNS). After glutamate is released and interacts with postsynaptic receptors, excitatory stimuli disappear with neurotransmitter removal from the synaptic cleft by glutamate reuptake transporters. Excitatory amino acid transporter 2 is the most abundant of these transporters. Excitotoxicity due to hyperactivation of glutamatergic receptors may result from increased synaptic levels of the neurotransmitter or greater sensitivity to glutamate in the postsynaptic terminal.100 AMPA receptors constitute one of the most important mediators of glutamatergic neurotransmission in the CNS. The permeability of these receptors to calcium is largely determined by a subunit protein called GluR2. Motor neurons are especially sensitive to excitotoxicity secondary to AMPA receptor activation, given the low expression of GluR2 and calcium buffering proteins in these cells.101,102 Several studies have suggested that excitotoxicity plays a role in the pathogenesis of ALS. Some patients with the disease have displayed high CSF glutamate levels and decreased expression of excitatory amino acid transporter 2 in the affected areas of the CNS.103,104 Other studies have shown cortical hyperexcitability in presymptomatic stages of the disease and abnormal calcium permeability in the AMPA receptors.105,106 Whether excitotoxic damage is a primary pathogenic mechanism or the consequence of a failure of neuronal homeostasis, it seems clear that once symptoms are established, alterations in neuronal homeostasis contribute to disease progression.
Neuroinflammation and the role of glial cellsAlthough motor neuron degeneration and loss is the main finding in patients with ALS, the condition has also been associated with activation of a neuroinflammatory response. Histological studies of the spinal cord of patients with ALS have found activated microglia and lymphocytic infiltrates.107 This inflammatory response contributes to the progressive degeneration and phenotypic alteration of motor neurons, starting with the first manifestations of the disease.108,109 Studies of biological samples from patients with ALS have revealed inflammatory mediators (increased CSF levels of interleukin-8) and parameters suggestive of immune response activation in peripheral blood.110
Astrocytes are glial cells which are closely associated with motor neurons. According to the literature, astrocytes can also induce motor neuron degeneration. Mutant SOD1 astrocytes have been shown to secrete a series of inflammatory mediators, including prostaglandin E2, leukotriene B4, and nitric oxide, which damage motor neurons.111 Subsequent studies have found the astrocytes of sALS patients not carrying SOD1 mutations to have a similar toxic effect.112 Oligodendrocytes have also been reported to play a role in the pathogenesis of motor neuron diseases, although to a lesser extent. In addition to myelinating CNS nerve fibres, oligodendrocytes contribute to regulating axonal metabolism, reducing lactic acid concentration in neurons by means of lactate transporter MCT1.113 According to several researchers, MCT1 loss is toxic to motor neurons. Likewise, decreased expression of the transporter has been observed in patients with ALS and mouse models of mutant SOD1.114
ConclusionsThere is mounting evidence that ALS is not a disease, but rather a clinical syndrome characterised by upper and lower motor neuron degeneration and distinctive clinical symptoms. A small percentage of cases of ALS are associated with mutations in specific genes; alterations in these genes lead to motor neuron degeneration. Although the aetiopathogenesis of sporadic forms of ALS is yet to be fully understood, it has been hypothesised that it may involve alterations in several cell pathways, including gene processing, proteostasis and protein aggregation, oxidative stress, and alterations in the neuronal microenvironment. These forms of ALS are probably due to the interaction between environmental factors and a genetic predisposition to the condition. Future research will contribute to our understanding of ALS pathogenesis, enabling the development of new therapeutic strategies.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Riancho J, Gonzalo I, Ruiz-Soto M, Berciano J. ¿Por qué degeneran las motoneuronas? Actualización en la patogenia de la esclerosis lateral amiotrófica. Neurología. 2019;34:27–37.
