






Desminopathies are a clinically heterogeneous group of myopathies, with common histological findings in muscle biopsy. Clinically, they usually present with distal and/or proximal muscle weakness often associated with cardiomyopathy. We present 8 patients from 3 unrelated families manifesting isolated respiratory insufficiency without skeletal muscle weakness or heart disease, because of a mutation in the DES gene.
MethodsClinical and demographic data were acquired from medical records. Muscle MRI studies were performed in 6 patients. A muscle biopsy study was performed in the index case from each family.
ResultsIsolated restrictive respiratory dysfunction was observed in all symptomatic patients, with 2 requiring non-invasive ventilation. Three patients were asymptomatic at the time of the study. None of the patients presented skeletal muscle weakness or heart disease, even after 20 years of disease progression. Muscle MRI showed a common pattern with predominant involvement of the semitendinosus muscle. Muscle biopsy showed patches of cytoplasmic inclusions corresponding to desmin aggregates. The genetic study showed heterozygous presence of the p.Arg415Trp mutation in the DES gene in all patients.
ConclusionsWe present 5 patients carrying a p.Arg415Trp mutation in the DES gene, manifesting as isolated restrictive respiratory insufficiency without associated skeletal muscle weakness or heart disease. These cases represent a new phenotype associated with DES mutations, thus suggesting that desminopathy should be considered in the diagnostic workup of patients presenting isolated respiratory failure.
Las desminopatías son un grupo de miopatías clínicamente heterogéneas, que comparten hallazgos histológicos comunes en la biopsia muscular. Clínicamente, suelen presentarse con una debilidad muscular distal y/o proximal que a menudo se asocia con una miocardiopatía. Presentamos ocho pacientes de tres familias no emparentadas que se manifiestan como una insuficiencia respiratoria aislada sin debilidad musculoesquelética ni cardiopatía asociada como consecuencia de una mutación en el gen DES.
MétodosLos datos clínicos y demográficos se obtuvieron de las historias clínicas. Se realizó estudio de resonancia magnética muscular en seis pacientes. En los casos índices de cada familia se realizó una biopsia muscular.
ResultadosSe observó una disfunción respiratoria restrictiva aislada en todos los pacientes sintomáticos, requiriendo VNI en dos de ellos. Tres pacientes estaban asintomáticos en el momento del estudio. Ninguno de los pacientes presentaron debilidad musculoesquelética o cardiopatía, incluso después de 20 años de evolución de la enfermedad. La RM muscular mostró un patrón común con afectación predominante del músculo semitendinoso. La biopsia muscular mostró parches de inclusiones citoplasmáticas correspondientes a agregados de desmina. El estudio genético mostró la misma mutación p.Arg415Trp en heterocigocis en el gen DES en todos los pacientes estudiados.
ConclusionesPresentamos cinco pacientes portadores de la mutación p.Arg415Trp en el gen DES que se manifiesta únicamente como una insuficiencia respiratoria restrictiva aislada sin debilidad musculoesquelética asociada ni cardiopatía. Estos casos representan un nuevo fenotipo asociado con mutaciones DES, lo que sugiere que las desminopatías deben ser consideradas en el proceso diagnóstico de los pacientes con una insuficiencia respiratoria aislada.
Desminopathies are a clinically heterogeneous group of diseases pathologically characterized by the presence of intracytoplasmic desmin-immunoreactive deposits in skeletal and cardiac muscles, seen as granulofilamentous material at the ultrastructural level.1,2
Desminopathies are caused by mutations in the DES gene, which codes for the protein desmin, the main intermediate filament in skeletal, cardiac, and smooth muscle tissue. Its function is decisive in the interaction with other proteins to form the cytoskeletal network that maintains the relationship between the contractile apparatus and the other elements of the muscle fiber, playing an important role in maintaining cell integrity, force transmission, and mechanochemical signaling.3 Despite being an autosomal dominant myopathy, recessive cases or de novo desmin mutations can also be found.4–7
Phenotypically, desminopathies are characterized by progressive muscle weakness, predominantly affecting the lower limbs, especially in distal regions.8 However, proximal, truncal, bulbar, or facial involvement may also be present.9–11 In addition, cardiac conduction defects and/or cardiomyopathy are observed in a large proportion of patients. These symptoms can manifest at onset or during progression of the disease, either isolated or in combination.12,13 Respiratory dysfunction is also frequently observed during the course of the disease, usually in patients presenting skeletal muscle weakness.3,14,15 Isolated restrictive respiratory failure is not a common feature in the majority of muscular dystrophies or other myopathies and, to our knowledge, has not yet been reported in desminopathies.
We report 8 patients from 3 unrelated families affected by desminopathy presenting with isolated restrictive respiratory dysfunction, without muscle weakness or cardiopathy. These cases reveal a new phenotype associated with DES mutations and demonstrate the importance of muscle biopsy and other complementary tests to support the diagnosis.
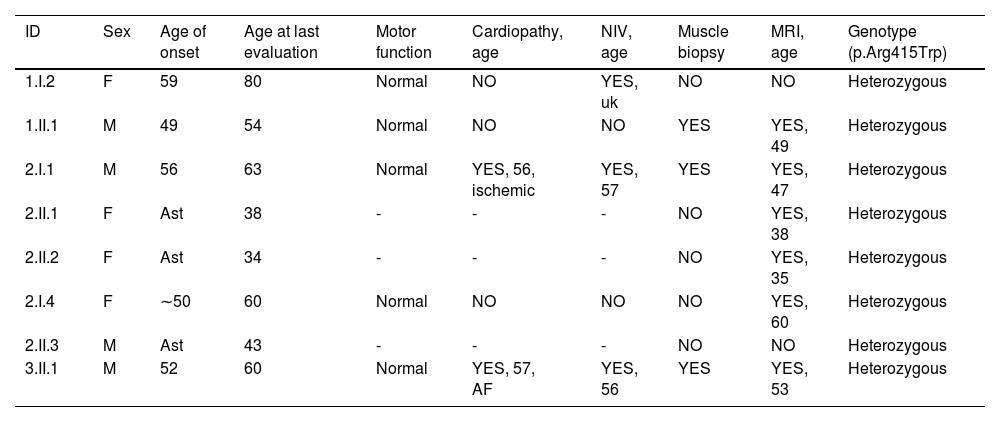
Materials and methodsClinical evaluationThe probands were three patients from three unrelated families, referred for respiratory dysfunction or a restrictive pattern in a respiratory function test. Patients from family 1 were evaluated at the neuromuscular diseases unit at Hospital de la Santa Creu i Sant Pau, whereas families 2 and 3 were studied at Hospital Universitari Parc Taulí. All 8 of the patients assessed, whether symptomatic or asymptomatic, were evaluated by a neurologist with experience in neuromuscular disorders. Subsequent studies included electrophysiological and imaging studies. Skeletal muscle biopsy was performed on the 3 index cases. Blood samples from each examined individual were collected for DNA extraction and genetic analysis. The clinical information of all patients was collected after obtaining informed consent, and the protocol was approved by the ethics committee at each center. Demographic and clinical characteristics are shown in Table 1.
Clinical and demographic characteristics of our series.
| ID | Sex | Age of onset | Age at last evaluation | Motor function | Cardiopathy, age | NIV, age | Muscle biopsy | MRI, age | Genotype (p.Arg415Trp) |
|---|---|---|---|---|---|---|---|---|---|
| 1.I.2 | F | 59 | 80 | Normal | NO | YES, uk | NO | NO | Heterozygous |
| 1.II.1 | M | 49 | 54 | Normal | NO | NO | YES | YES, 49 | Heterozygous |
| 2.I.1 | M | 56 | 63 | Normal | YES, 56, ischemic | YES, 57 | YES | YES, 47 | Heterozygous |
| 2.II.1 | F | Ast | 38 | - | - | - | NO | YES, 38 | Heterozygous |
| 2.II.2 | F | Ast | 34 | - | - | - | NO | YES, 35 | Heterozygous |
| 2.I.4 | F | ∼50 | 60 | Normal | NO | NO | NO | YES, 60 | Heterozygous |
| 2.II.3 | M | Ast | 43 | - | - | - | NO | NO | Heterozygous |
| 3.II.1 | M | 52 | 60 | Normal | YES, 57, AF | YES, 56 | YES | YES, 53 | Heterozygous |
F: female; M: male; Ast: asymptomatic; AF: atrial fibrillation; NIV: non-invasive ventilation; uk: unknown
Genetic studies of all index patients were performed with next-generation sequencing (NGS) technology, analyzing a panel of a total of 106 genes associated with myopathies. The list of genes analyzed is shown in supplementary Figure 1. In the affected and healthy relatives, the genetic study of the DES gene was carried out using Sanger sequencing.
Muscle biopsy analysisSkeletal muscle biopsy samples were obtained by the open procedure in the index cases of all 3 families as part of the diagnostic process. The sample was obtained from the right gastrocnemius muscle in family 1, from the right tibialis anterior in family 2, and from the left quadriceps in family 3. The samples were immediately frozen in liquid nitrogen–chilled isopentane and processed for routine histochemical reactions and immunohistochemistry for desmin, dystrophin, αB-crystallin, myotilin, and ubiquitin.
Muscle resonance imagingWhole-body muscle resonance imaging (MRI) was performed in all patients, with the exception of patients 1.I.2 and 2.II.3. In patient 3.II.1, MRI was performed only on the thigh. Patients were examined using a 1.5T MRI scanner as part of the routine workup at the participating centers. We analyzed whole-body or lower-limb MRI axial T1-weighted sequences and short-tau inversion recovery (STIR) sequences. Axial T1-weighted muscle images were scored with the semi-quantitative Mercuri scale modified by Fischer,16 as follows: 0, normal appearance; 1, mild involvement, less than 30% of individual muscle volumes; 2, moderate involvement, 30%-60% of muscle affected; 3, severe involvement, >60% of muscle affected; 4, end stage, all the muscle affected. For the STIR sequence, we recorded whether the muscles presented signal hyperintensity.
ResultsClinical features and outcomesFamily 1Patient 1.II.1 (Fig. 1A) is a 49-year-old white man, born to non-consanguineous parents, referred to our center due to a restrictive respiratory pattern in the pulmonary function test performed at his employer's annual health check-up. He did not report orthopnea or symptoms suggestive of nocturnal hypoventilation, nor muscle weakness, palpitations, or other neuromuscular or cardiac symptoms. Neurological examination revealed normal muscle strength in all muscles of all 4 limbs, with no associated muscular atrophies. The pulmonary function test showed a restrictive pattern with a decrease in forced vital capacity (FVC) (49%), forced expiratory volume in 1 second (FEV1) (45%), and maximal inspiratory pressure (MIP) (42%). Chest radiography and tomography studies showed no lung parenchymal abnormalities, and dynamic MRI showed diaphragmatic mobility within normal limits, although with a slight elevation of the left diaphragm. A 24-hour Holter monitoring study to identify arrhythmias or other cardiac conduction defects showed no abnormalities, whereas cardiac ultrasound showed mild hypertrophy of the left ventricle. The results of nerve conduction studies and needle electromyography (EMG) were normal. Serum creatine kinase (CK) levels were mildly elevated (223 IU/L; normal range, <170 IU/L), and the remaining blood biochemical analysis parameters were normal.

Pedigrees of family 1, 2, and 3. Closed symbols represent affected symptomatic family members, while semi-closed symbols represent asymptomatic carriers. The arrows indicate the index patients.
+: p.Arg415Trp DES allele; -: wild-type DES allele; ND: not done.
Pedigrees were slightly adapted for anonymization purposes.
Patient 1.I.2 was the deceased mother of patient 1.II.1. She had been evaluated in our center at the age of 71 years due to bilateral diaphragmatic paralysis with a restrictive respiratory pattern, requiring nocturnal non-invasive ventilation. At that time, the neurological examination and neurophysiological study findings were completely normal, and no underlying neuromuscular disease was suspected. The patient died at the age of 80 years due to respiratory failure, with no associated skeletal muscle weakness or cardiac involvement. Considering the pedigree, an autosomal dominant hereditary disease was suggested.
Family 2Patient 2.I.1 (Fig. 1B) is a 56-year-old white man born to non-consanguineous parents, who was evaluated 5-6 years ago due to exertional dyspnea. Bilateral diaphragmatic paralysis was observed. The patient also complained of muscle cramps in the lower limbs. At that time, the neurological examination revealed normal muscle strength without muscle atrophy. Nerve conduction and needle EMG study findings were normal. Serum CK levels were slightly elevated (263 IU/L), whereas the remaining blood biochemical analysis parameters were normal. Spirometry testing revealed decreased FVC (48%) and FEV1 (47%), which worsened in a decubitus position (FVC 35% and FEV1 27%), suggesting diaphragmatic dysfunction. Non-invasive respiratory support was required at the age of 57. Cardiological evaluation showed abnormal anterior wall motion due to a myocardial infarction at the age of 55. An extensive study to identify arrhythmias or heart conduction disorders showed no abnormalities. At the last evaluation, aged 63 years, the patient was able to walk without support and showed no muscle weakness.
Patients 2.II.1 and 2.II.2 were daughters of patient 2.I.1 and were considered asymptomatic at the time of the analysis. Their last evaluations were performed at the ages of 38 and 34 years, respectively. Neither presented muscle weakness, and respiratory function test results were normal. CK levels were also normal (81 IU/L and 51 IU/L, respectively).
Patient 2.I.4 (Fig. 1B) was the sister of patient 2.I.1. Since approximately 50 years of age, she had presented dyspnea with moderate exertion and morning headache, with no associated limb weakness. She was diagnosed at the age of 57, following her brother's diagnosis (patient 2.I.1). The patient presented no muscle weakness in neurological examination, and serum tests revealed slightly elevated CK levels (251 IU/L). Pulmonary function testing showed decreased FVC (69%), and a complete cardiological workup showed no alterations. At the last evaluation, at 60 years of age, the patient still had no evident muscle weakness.
Patient 2.II.3 was the son of patient 2.I.4. His last assessment was performed at the age of 43 years. The patient did not report any symptoms and neurological examination and pulmonary function test findings were normal. CK levels were slightly elevated (275 IU/L). This patient was considered asymptomatic at the time of the study.
Family 3Patient 3.II.1 (Fig. 1C) is a white man born to non-consanguineous parents, who was evaluated at 52 years of age due to progressive effort dyspnea and bilateral diaphragmatic paralysis of several years’ progression. At that time, neurological examination revealed normal muscle strength and no atrophy. Nerve conduction studies showed mild sensory axonal polyneuropathy in the lower limbs, and needle EMG shown motor units with myopathic pattern. CK levels were normal (86 IU/L). Spirometry showed a severe decrease in FVC (27%), requiring non-invasive respiratory support at 53 years of age. Cardiological evaluation revealed moderate ventricular hypertrophy and atrial fibrillation at 57 years of age. At the last evaluation, when the patient was 60 years old, no muscle weakness was observed.
Muscle magnetic resonance imagingMuscle MRI was performed in all patients with confirmed genetic diagnosis, except for patients 1.I.2 and 2.II.3. The studies showed a common pattern in all patients, consisting predominantly of semitendinosus muscle involvement. In patient 1.II.1, from family 1, the study was performed at the age of 50 years and revealed severe selective fatty replacement of both semitendinous muscles on T1-weighted images (Mercuri 4), with normal appearance of the remaining muscles (Fig. 2A). In the other symptomatic patient in family 1, patient 1.I.2, no MRI study was performed. In family 2, patients 2.I.1 (Fig. 2B) and 2.I.4 underwent MRI studies at 57 and 60 years of age, respectively; in addition to the almost selective involvement of both semitendinosus muscles (Mercuri 3-4), the study revealed slight fatty replacement of the sartorius (Mercuri 2), tensor fasciae latae (Mercuri 1), and gluteus muscles (Mercuri 2), as well as both the medial and lateral heads of the gastrocnemius (Mercuri 3). Two asymptomatic patients from family 2, patients 2.II.1 and 2.II.2 (Fig. 2C), underwent MRI at 35 and 38 years of age, respectively, presenting very mild fatty infiltration in both semitendinosus muscles (Mercuri 1) and in paraspinal muscles (Mercuri 2). In the other asymptomatic patient from family 2, patient 2.II.3, no MRI study was performed. In family 3, muscle MRI was performed in patient 3.II.1 at 53 years of age, revealing fatty replacement in both gluteus (Mercuri 3) and semitendinosus muscles (Mercuri 4). STIR images were normal in all patients, with no areas of signal hyperintensity.
Morphological analysis, 2.I.1 (B), and 2.II.2 (C).")
Muscle biopsy (fig. 3) was performed in all 3 index cases. All biopsies revealed marked variation in the size of the muscle fibers and an increased number of internal nuclei. Rimmed vacuoles were observed in some fibers. The most relevant finding was the presence of deposits of eosinophilic material with hematoxylin-eosin (HE) stain and dark green with modified Gömöri trichrome stain, located under the sarcolemma or within the cytoplasm in several fibers. These areas occupied by the deposits were devoid of oxidative enzyme activity. The deposits were strongly immunoreactive for desmin, myotilin, and p62.
 from family 1. Hematoxylin-eosin staining (A) shows increased fiber-size variation, internalized myonuclei, and an increase in endomysial connective tissue. Many fibers contain deposits of eosinophilic material, seen as bluish-red with the modified Gömöri trichrome staining (B). In the NADH (C) and COX reactions (D), the areas occupied by the deposits are devoid of oxidative enzyme activity and give the appearance of rubbed-out fibers. These areas are immunoreactive for desmin (E-F).")
Histological analysis and immunohistochemistry of skeletal muscle biopsy of the index patient (1.II.1) from family 1. Hematoxylin-eosin staining (A) shows increased fiber-size variation, internalized myonuclei, and an increase in endomysial connective tissue. Many fibers contain deposits of eosinophilic material, seen as bluish-red with the modified Gömöri trichrome staining (B). In the NADH (C) and COX reactions (D), the areas occupied by the deposits are devoid of oxidative enzyme activity and give the appearance of rubbed-out fibers. These areas are immunoreactive for desmin (E-F).
The genetic study detected heterozygous presence of the missense mutation c.1243C>T, p.Arg415Trp (NM_001927.4) in exon 6 of the DES gene in all affected members of the 3 families, and also in the 3 young asymptomatic individuals.
The variant is located in a well-conserved domain, in up to 10 species. A sequence-based prediction of impact of the missense mutation p.Arg415Trp on protein function was performed using different computational approaches assessing sequence conservation; for instance, CADD,17 REVEL,18 and SIFT;19 sequence and structural features were studied with PolyPhen-220 (Harvard University, Massachusetts, US) and MutationTaster (Berlin Institute of Health, Berlin, Germany).
The scores obtained with the mentioned mutation prediction tools for the mutation p.Arg415Trp were the following: CADD score: 25.3 (a score of 20 or higher indicates that the variant is predicted to be among 1% of the most deleterious possible variants in the human genome); REVEL score: 0.811 (scores range from 0 to 1 and variants with higher scores are predicted to be more likely to be pathogenic); SIFT score: 0 (the score is the normalized probability that the amino acid change is tolerated, so scores nearer zero are more likely to be deleterious); PolyPhen-2 prediction: probably damaging with a score of 1.000 (the PolyPhen-2 score ranges from 0.0 [tolerated] to 1.0 [deleterious]); mutation taster prediction: disease-causing.21
The mutation p.Arg415Trp presents an extremely low frequency in databases such as EXAC (3/119 160 [0.000025]) and gnomAD (2/31 370 [0.000064]); it has been reported to ClinVar and LOVD and classified as a variant of uncertain significance.
DiscussionWe report 5 patients from 3 unrelated families with presumed autosomal dominant inheritance presenting with isolated restrictive respiratory dysfunction, without muscle weakness or associated cardiomyopathy as a result of the same p.Arg415Trp mutation in the DES gene. Muscle biopsy performed in the proband from each family showed cytoplasmic aggregates immunoreactive for desmin. Respiratory dysfunction was detected during the 6th decade of life in all symptomatic cases. Three additional mutation carriers under the age of 50 years, from family 2, were asymptomatic, although slight alterations were observed in the muscle MRI study. This clinical presentation with isolated respiratory dysfunction broadens the phenotypic variability of desminopathies and highlights the importance of evaluating this type of patients in specialized neuromuscular diseases units. Better understanding of the phenotypic variability of this disease is especially important today, when new experimental therapies are being developed.
Though the main symptom is muscle weakness, desminopathies can present with cardiac involvement in the form of conduction defects and/or cardiomyopathy, even in the absence of skeletal muscle weakness.8 The age of onset of the disease is usually 30 years, being earlier in recessive cases and in those with isolated cardiac involvement.15 In our 5 symptomatic patients, symptoms began at over 50 years of age, isolated respiratory failure without muscle weakness or cardiac involvement, symptoms that did not appear during follow-up. In the 3 asymptomatic carriers, the last evaluation was performed at the ages of 38, 34, and 43 years. These data suggest that onset in patients carrying the p.Arg415Trp mutation occurs around the 6th decade of life, which would explain why the asymptomatic carriers in our series have not yet developed the disease, despite subtle muscle involvement on MRI. This reinforces the importance of regular follow-up and monitoring of these patients.
The presence of a restrictive respiratory pattern of unknown cause should prompt the search for a neuromuscular disorder.22,23 Although isolated respiratory involvement without associated muscle weakness is uncommon in neuromuscular diseases, it may appear at onset, so differential diagnosis is essential. For example, approximately 3% of patients with amyotrophic lateral sclerosis present in this way;24 respiratory failure can also be the initial manifestation in up to 14% of patients with myasthenia gravis.25 The results of the neurophysiological study, muscle MRI, and muscle biopsy confirmed primary muscle involvement in our patients. Several myopathies may develop with early respiratory involvement, even as the initial symptom of the disease, such as late onset Pompe disease, although this is often associated with progressive muscle weakness.26 Respiratory dysfunction is also a common feature in Steinert disease (in fact, non-invasive ventilation is necessary in many patients), although muscle weakness, cardiac involvement, and myotonia are also symptoms of this disease.27 Similarly, respiratory involvement may be present in late stages of limb-girdle muscular dystrophies such as sarcoglycanopathies, calpainopathies, or fukutinopathies, when muscle weakness is extensive.28 Hereditary myopathy with early respiratory failure, caused by mutations in TTN, is a myopathy that could present with a similar phenotype as in our patients, despite later development of both proximal and distal limb muscle weakness. Furthermore, these patients also share predominant semitendinosus involvement in the muscle MRI and show similar findings in the muscle biopsy.29 In our patients, genetic analysis ruled out possible mutations in the TTN gene. In desminopathies, respiratory involvement may be present in 26% of patients over the course of the disease, although it is normally associated with muscle weakness and/or cardiomyopathy.2 The diaphragm, unlike other skeletal muscles, functions in an environment in which forces can be transmitted both longitudinally and transversely during each respiratory cycle; desmin is the only known molecule with a dual orientation, and therefore serves as a viscoelastic element that dissipates mechanical energy on both planes. Reflecting on this critical significance of desmin in respiratory function, its content in the diaphragm is 38% higher than in the biceps femoris muscle.30 This highlights the important role of desmin in the diaphragm. Our patients presented with severe restrictive respiratory involvement, with some of them requiring early non-invasive ventilation at ages 57 and 53, with this dysfunction even being the cause of death in one patient. Respiratory involvement is isolated, without muscle weakness or associated cardiomyopathy, despite disease progression times of over 20 years and patient age older than 80 years, as occurs in some of our patients.
The DES gene comprises 9 exons that code for the protein desmin. This protein is made up of a highly conserved head and tail sequence and 4 alpha-helical domains: 1A, 1B, 2A, and 2B. Most mutations are missense type and occur in the 2B domain, with these patients most frequently presenting a neurological phenotype with or without associated heart disease, whereas patients with isolated heart disease usually present mutations in the head or tail domain.31 According to a recent study, pathogenic variants located in the 1B segment and in the tail domain are more likely to present as early-onset cardiomyopathy, compared to patients with variants in other segments;32 in addition, isolated cardiac or respiratory involvement as symptom of onset may be present in up to 65% of patients with mutations in the head or tail domain.33 The patients reported in this work carry the mutation c.1243C>T, p.Arg415Trp located in exon 6, which is located in the tail domain. The population prevalence of this mutation is extremely low, and most predictors classify the mutation as deleterious. The characteristic muscle MRI pattern, together with the presence of several features of desminopathies on muscle biopsy, such as myofibrillar disintegration and deposits of eosinophilic material corresponding to accumulations of desmin, support the pathogenicity of the variant found in the DES gene and help confirm the diagnosis of desminopathy in these patients.
In conclusion, we present 5 patients from 3 unrelated families affected by a desminopathy secondary to a novel DES mutation. These patients presented with isolated restrictive respiratory involvement, without associated muscle weakness or myocardiopathy, expanding the phenotypic spectrum of desminopathies, with this diagnosis being a possible cause of isolated respiratory involvement.
Funding statamentJorge Alonso-Pérez and Álvaro Carbayo are supported by the Rio Hortega grant (CM19/00178 and CM21/00057, respectively). Acción Estratégica de Salud (EAS), Instituto de Salud Carlos III (Spain). Lidia Gonzalez-Quereda and Pia Gallano are supported by Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III, Spain and ERDF funds “A way to make Europe”/“Investing in your future”) under Grant PI18/01585. Montse Olivé is supported by Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III, Spain and EDRF funds “A way to make Europe”/“Investing in your future”) under Grant PI21/01621.
Financial disclosuresAll authors have no relevant disclosures to report.
Ethics approvalThe study was approved by the ethics committee of Sant Pau Hospital.
Data sharingThe data that support the findings of this study are available upon reasonable request from the corresponding author.
Conflicts of interestAll authors report no conflicts of interest.
J. Alonso-Pérez, A. Carbayo, J. Diaz-Manera, M. Olivé, and R. Rojas-García are members of the European Reference Network for Neuromuscular Diseases.
