




Huntington disease (HD) is a hereditary neurodegenerative disorder. Thanks to predictive diagnosis, incipient clinical characteristics have been described in the prodromal phase.
ObjectiveTo compare performance in cognitive tasks of carriers (HDC) and non-carriers (non-HDC) of the huntingtin gene and to analyse the variability in performance as a function of disease burden and proximity to the manifest stage (age of symptom onset).
MethodA sample of 146 participants in a predictive diagnosis of HD programme were divided into the HDC (41.1%) and non-HDC groups (58.9%). Mathematical formulae were used to calculate disease burden and proximity to the manifest stage in the HDC group; these parameters were correlated with neuropsychological performance.
ResultsSignificant differences were observed between groups in performance on the Mini–Mental State Examination (MMSE), Stroop-B, Symbol-Digit Modalities Test (SDMT), and phonological fluency. In the HDC group, correlations were observed between disease burden and performance on the MMSE, Stroop-B, and SDMT. The group of patients close to the manifest stage scored lowest on the MMSE, Stroop-B, Stroop-C, SDMT, and semantic verbal fluency. According to the multivariate analysis of covariance, the MMSE effect shows statistically significant differences in disease burden and proximity to onset of symptoms.
ConclusionsMembers of the HDC group close to the manifest phase performed more poorly on tests assessing information processing speed and attention. Prefrontal cognitive dysfunction appears early, several years before the motor diagnosis of HD.
La enfermedad de Huntington (EH) es un trastorno neurodegenerativo y hereditario, gracias al diagnóstico predictivo se han descrito características clínicas incipientes en la fase prodrómica.
ObjetivoComparar la ejecución en tareas cognitivas de portadores (PEH) del gen de la huntingtina y no portadores (NPEH) y observar la variabilidad en la ejecución, dependiendo de la carga de la enfermedad y cercanía a la etapa manifiesta (edad de inicio de síntomas).
Método146 participantes de un Programa de Diagnóstico Predictivo de EH (PDP-EH) se dividió en PEH (41.1%) y NPEH (58.9%). Mediante fórmulas matemáticas se obtuvo la carga de enfermedad y cercanía a la etapa manifiesta en el grupo PEH y se correlacionó con la ejecución neuropsicológica.
ResultadosSe observaron diferencias significativas entre grupos en MMSE, Stroop-B, SDMT y fluidez fonológica. En el grupo PEH se observaron correlaciones entre la carga de enfermedad con el MMSE, Stroop-B y SDMT. El grupo “Cerca” a la etapa manifiesta es el que obtiene la puntuación más baja en el MMSE, Stroop-B, Stroop-C, SDMT y fluidez verbal semántica. De acuerdo al MANCOVA el efecto MMSE evidencia diferencias estadísticamente significativas entre carga de la enfermedad y cercanía de inicio de síntomas.
ConclusionesSe observa un nivel menor de desempeño en el grupo PEH con probabilidad de inicio cercano de la fase manifiesta, en pruebas que evalúan velocidad de procesamiento y atención. La disfunción cognitiva prefrontal se altera de manera precoz, varios años antes del diagnóstico motor de la EH.
Huntington disease (HD) is a hereditary neurodegenerative disorder that follows an autosomal dominant inheritance pattern. It is characterised by motor, psychiatric, and cognitive alterations. Its prevalence amounts to 10 cases per 100 000 population in Western countries.1 HD is caused by a CAG triplet repeat expansion in the gene encoding huntingtin (HTT), at locus 4p16.32,3; 4 repeat ranges are considered: the < 27 repeats range is associated with normal phenotype; 27–35 with intermediate alleles; 36–39 with incomplete penetrance; and > 40 with complete penetrance. These ranges explain the differences in the outcomes of presymptomatic patients.4
Clinical symptoms of HD generally manifest in the fourth decade of life, with a progression time between 15 and 20 years.3,5 Currently, no curative treatment or disease-modifying therapy exists.6 Progression of HD may be divided into 2 periods: presymptomatic and symptomatic. The presymptomatic period includes 2 stages: the presymptomatic stage, in which carriers are not clinically distinguishable from healthy subjects, and the prodromal stage, characterised by mild psychiatric, motor, and cognitive symptoms.7 The symptomatic stage formally starts when clinical motor diagnosis is established; in other words, when the patient presents motor changes that may include such abnormal movements as chorea, bradykinesia, dystonia, and lack of coordination, with a likelihood ≥ 99% that the cause is HD.8,9
According to Reilmann et al.,7 the prodromal stage is especially interesting for clinical research as it is the ideal time to test treatments that may delay the onset and modify the progression of the disease, with the smallest possible number of adverse effects. Thanks to diagnostic tests for HD, early clinical characteristics have been described that manifest in the prodromal stage and are potential clinical markers of the disease.10 Some of these markers are related to cognitive functions. When comparing performance in cognitive tasks in individuals at risk of developing HD, researchers have observed that carriers of the mutation present poorer cognitive performance than non-carriers, mainly in tasks evaluating information processing speed, executive function, attention, episodic memory, and visuospatial skills.9,11,12
Furthermore, there is an inverse correlation between the number of repeats and the age of disease onset: the greater the number of repeats, the earlier symptoms present. In other words, larger repeat expansions are associated with a higher probability of brain tissue degeneration, motor and cognitive symptoms, and the loss of functional capacity for activities of daily living manifesting at younger ages.13
Some authors have analysed the association between the progression of cognitive symptoms in carriers of the mutation causing HD and the genetic burden of the disease using mathematical formulas that consider individual genetic differences.14 One of these formulas for quantifying disease burden was developed by Penney et al.,15 who proposed the formula: disease burden = current age × (CAG – 35.5). This formula has been used in several studies.7,14–16
Campodonico et al.17 designed a formula to predict the age of onset of motor symptoms considering the number of CAG triplet repeats and the parental age at onset, among others. Brandt et al.18 updated the 2-variable formula, finding that the modified algorithm accounted for 62% of variance in time to onset.
Our study analyses the following hypotheses: a) mutation carriers will show greater variability in cognitive task performance than non-carriers; and b) among carriers, variability in cognitive task performance will increase in line with disease burden and proximity to the symptomatic stage (age of symptom onset).
MethodsThe genetics department of the Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez invites subjects at risk of developing HD to participate in a Huntington Disease Predictive Testing Programme (HDPTP).19 This programme is approved by the institute’s research and ethics committee and complies with the recommendations of the International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea.20
Between 2002 and 2019, a total of 189 participants gave verbal and written informed consent to participate in the HDPTP. A multidisciplinary team (clinical geneticist, psychiatrists, neurologists, neuropsychologists, social worker) performed both clinical and cognitive evaluations of all participants. Exclusion criteria included the presence of apathy, anxiety, or depression; the detected cases were referred to the psychiatry clinic and their participation was postponed until they showed improvement.
The results obtained in the different assessments were recorded in a database using the SPSS software (version 22.0). We applied the following exclusion criteria: history of risk of suicide or any untreated psychiatric disease, incomplete neuropsychological tests, unknown results of molecular testing, and clear symptoms of the disease or of any other neurological condition. Thirty patients were excluded due to incomplete cognitive tests, and another 13 due to unknown genetic study results. A total of 146 participants were included in the study and grouped according to their molecular testing results.
For practical purposes, we categorised the sample in accordance with previous studies,7,14,15,18 establishing 2 groups: mutation carriers, referring to individuals with > 36 CAG triplet repeats in the huntingtin gene; and non-carriers, referring to those with ≤ 36 CAG triplet repeats in both alleles.
ToolsThe HDPTP is run over several sessions, one of which focuses on the neurological and neuropsychological assessment of participants. In that session, patients complete a questionnaire gathering sociodemographic and clinical data, and a neurological assessment is performed using the Unified Huntington’s Disease Rating Scale.19 Cognitive assessment was performed using the Mini–Mental State Examination (MMSE)21 and neuropsychological tests standardised for the Mexican population22 and focused on assessing executive functions, as in other studies of people at risk of developing HD.23–26 The Stroop Color-Word Interference Test measures executive performance, and comprises 3 tasks: word reading (Stroop-A), colour naming (Stroop-B), and word-colour interference (Stroop-C). The first 2 parts have been used in HD to assess information processing speed, whereas the interference task has been used to assess cognitive function, and specifically inhibition.
The Symbol Digit Modalities Test (SDMT) is used to assess information processing speed.
Semantic verbal fluency in the categories “animals” and “fruits” and phonological verbal fluency with the letters “F” and “A” assess executive function, and cognitive flexibility in particular.22
Data analysisThis study is based only on the demographic and cognitive data obtained in the HDPTP. We performed a descriptive analysis of the demographic data, and used the chi-square test to compare categorical variables and ANOVA to compare continuous variables. We used the Shapiro-Wilk test to analyse normal distribution of cognitive data. Specific neuropsychological scores (dependent variables) were compared using one-way ANOVA, with diagnostic group as the independent variable. Given the number of univariate comparisons, the threshold for significance was set at P < .01 to minimise the probability of both type I and type II errors.
For each participant, we determined the number of CAG triplet repeats. Disease burden was calculated using the following formula: (CAGn − 35.5) × age. The values obtained ranged between 30 and 957 (mean: 292.52 [SD: 127.57]).
We subsequently used the formula proposed by Brandt et al.18 to classify mutation carriers into 3 groups: 1) the far-from-onset group, ie, individuals in whom cognitive symptom onset and diagnosis are expected to occur in more than 15 years; 2) the intermediate group, in whom onset and diagnosis are expected to occur in 9–15 years; and 3) the close-to-onset group, for whom cognitive symptom onset and diagnosis are expected within 9 years.
Lastly, in the multivariate analysis (MANCOVA), we used disease burden and proximity to the symptomatic stage (age at symptom onset) as covariates of neuropsychological scores. We calculated the Wilks’ lambda and the effect size, considering values of 0.04 as minimal, 0.25 as moderate, and 0.64 as large (η2).
ResultsOf 146 participants, 60 (41.1%) were classed as carriers and 86 (58.96%) as non-carriers. The mean number of CAG triplet repeats was 43.95 (SD: 3.3; range: 37–53) in carriers and 20.4 (SD: 3.4; range: 15–29) in non-carriers.
We observed a predominance of women in both groups (56.7% in carriers and 61.1% in non-carriers). Mean age was 35.9 years in carriers and 38 years in non-carriers. The level of schooling was similar in both groups (carriers: 14.5 years; non-carriers: 14.2 years). We identified no significant differences between groups in any demographic variable.
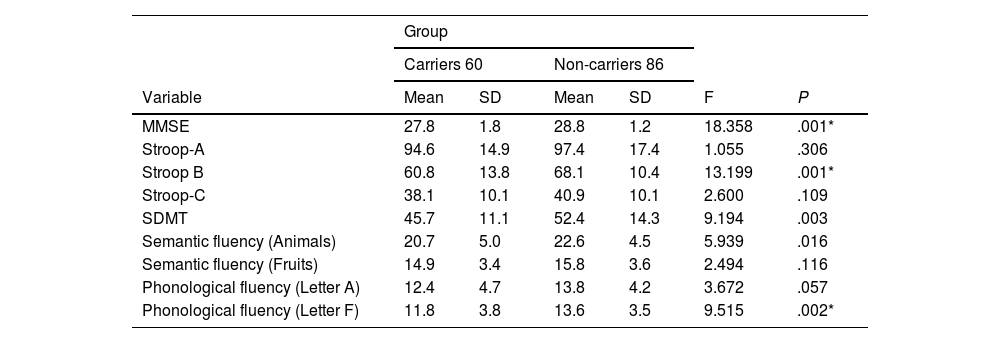
With regard to the comparison of cognitive performance between groups, we observed statistically significant differences in the MMSE, Stroop-B, SDMT, and phonological fluency (letter F) tests (Table 1).
Cognitive performance in carriers and non-carriers of the Huntington disease mutation.
| Group | ||||||
|---|---|---|---|---|---|---|
| Carriers 60 | Non-carriers 86 | |||||
| Variable | Mean | SD | Mean | SD | F | P |
| MMSE | 27.8 | 1.8 | 28.8 | 1.2 | 18.358 | .001* |
| Stroop-A | 94.6 | 14.9 | 97.4 | 17.4 | 1.055 | .306 |
| Stroop B | 60.8 | 13.8 | 68.1 | 10.4 | 13.199 | .001* |
| Stroop-C | 38.1 | 10.1 | 40.9 | 10.1 | 2.600 | .109 |
| SDMT | 45.7 | 11.1 | 52.4 | 14.3 | 9.194 | .003 |
| Semantic fluency (Animals) | 20.7 | 5.0 | 22.6 | 4.5 | 5.939 | .016 |
| Semantic fluency (Fruits) | 14.9 | 3.4 | 15.8 | 3.6 | 2.494 | .116 |
| Phonological fluency (Letter A) | 12.4 | 4.7 | 13.8 | 4.2 | 3.672 | .057 |
| Phonological fluency (Letter F) | 11.8 | 3.8 | 13.6 | 3.5 | 9.515 | .002* |
MMSE: Mini–Mental State Examination; SD: standard deviation; SDMT: Symbol Digit Modalities Test.
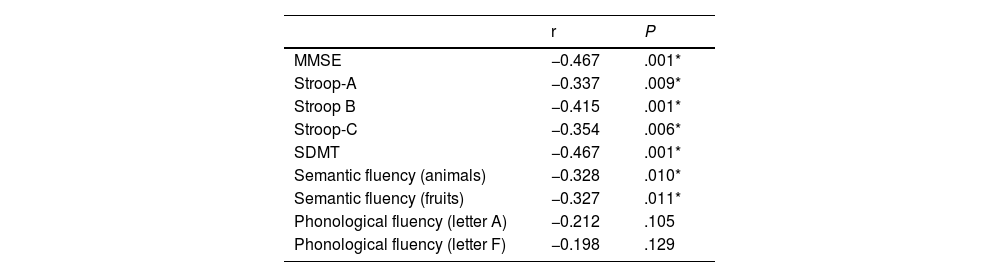
Table 2 shows the correlations between disease burden and performance in the different neuropsychological tests. The strongest associations were identified with the MMSE, Stroop (A, B, and C) tests, SDMT, and semantic fluency tests (animals and fruits), showing an inverse correlation between disease burden and the scores obtained in neuropsychological tests, which suggests that greater disease burden is associated with poorer test performance.
Pearson correlation coefficient between disease burden and neuropsychological performance in the group of carriers of Huntington disease mutation.
| r | P | |
|---|---|---|
| MMSE | −0.467 | .001* |
| Stroop-A | −0.337 | .009* |
| Stroop B | −0.415 | .001* |
| Stroop-C | −0.354 | .006* |
| SDMT | −0.467 | .001* |
| Semantic fluency (animals) | −0.328 | .010* |
| Semantic fluency (fruits) | −0.327 | .011* |
| Phonological fluency (letter A) | −0.212 | .105 |
| Phonological fluency (letter F) | −0.198 | .129 |
MMSE: Mini–Mental State Examination; SDMT: Symbol Digit Modalities Test.
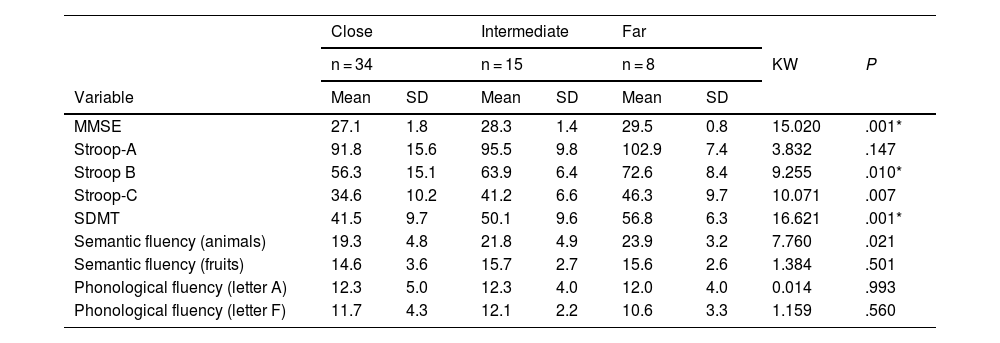
When categorising the carrier group according to the proximity to diagnosis, we observed a far-from-onset group (n = 8), an intermediate group (n = 15), and a close-to-onset group (n = 34). Age at onset of the parent who developed HD was not available in 3 cases, and therefore we were only able to categorise 57 of the 60 carriers. The post hoc Kruskal-Wallis test revealed the following findings: for the MMSE, the close-to-onset group scored the lowest. This same tendency was observed in the Stroop-B, Stroop-C, SDMT, and semantic fluency (animals) tests (Table 3).
Comparison by probability of close proximity to onset (time to diagnosis of motor symptoms).
| Close | Intermediate | Far | ||||||
|---|---|---|---|---|---|---|---|---|
| n = 34 | n = 15 | n = 8 | KW | P | ||||
| Variable | Mean | SD | Mean | SD | Mean | SD | ||
| MMSE | 27.1 | 1.8 | 28.3 | 1.4 | 29.5 | 0.8 | 15.020 | .001* |
| Stroop-A | 91.8 | 15.6 | 95.5 | 9.8 | 102.9 | 7.4 | 3.832 | .147 |
| Stroop B | 56.3 | 15.1 | 63.9 | 6.4 | 72.6 | 8.4 | 9.255 | .010* |
| Stroop-C | 34.6 | 10.2 | 41.2 | 6.6 | 46.3 | 9.7 | 10.071 | .007 |
| SDMT | 41.5 | 9.7 | 50.1 | 9.6 | 56.8 | 6.3 | 16.621 | .001* |
| Semantic fluency (animals) | 19.3 | 4.8 | 21.8 | 4.9 | 23.9 | 3.2 | 7.760 | .021 |
| Semantic fluency (fruits) | 14.6 | 3.6 | 15.7 | 2.7 | 15.6 | 2.6 | 1.384 | .501 |
| Phonological fluency (letter A) | 12.3 | 5.0 | 12.3 | 4.0 | 12.0 | 4.0 | 0.014 | .993 |
| Phonological fluency (letter F) | 11.7 | 4.3 | 12.1 | 2.2 | 10.6 | 3.3 | 1.159 | .560 |
Values are expressed as means and standard deviations.
Close: expected diagnosis of Huntington disease within 9 years; intermediate: expected diagnosis of Huntington disease in 9–15 years; far: expected diagnosis of Huntington disease in more than 15 years.
KW: Kruskal-Wallis test; MMSE: Mini–Mental State Examination; SD: standard deviation; SDMT: Symbol Digit Modalities Test.
To determine the effect of disease burden and proximity (time) to onset of motor symptoms, we use a multivariate analysis (MANCOVA) for each statistically significant variable.
Fig. 1 shows the results of the MANCOVA, with the scores obtained in the neuropsychological assessment. In the analysis with disease burden and proximity to symptom onset as covariates, we observed statistically significant differences with the neuropsychological scores, with Wilks’ lambda of 0.736 (F[5, 135] = 3,58; P < .001) and a large effect size (η2 = 0.998; r = 0.14). A multiple regression analysis (r = 0.226; F = 4.905; P = .009) revealed that the MMSE score showed statistically significant differences between disease burden and proximity to symptom onset (F[1,135] = −3.116; P = .002), with a large effect size (η2 = 0.871). This means that greater proximity to symptom onset is associated with poorer performance in the MMSE among mutation carriers. Furthermore, we observed a tendency (r = 0.149; F = 2.874; P = .060) that revealed that the Stroop-B score showed statistically significant differences between disease burden and proximity to symptom onset (F[1,135] = −2.397; P = .018), with a large effect size (η2 = 0.663). Carriers presented poorer performance when symptom onset was closer, unlike non-carriers.
Discussion
This is the first study in a Mexican population of participants in a predictive testing programme for HD. The cognitive results are consistent with several previous findings reported in the international literature.14,23–26
In accordance with the cognitive results, we observed that the mean MMSE scores of both groups were within normal ranges: no participant scored below 23 points, which is the cut-off point for cognitive impairment in Spanish-speaking populations.27 Several studies report that carriers and non-carriers of the HD mutation present similar cognitive performance, within normal ranges.27,28 However, our results show statistically significant differences between groups in total MMSE score, as well as a correlation between disease burden and the MMSE score among carriers. These changes in overall cognitive performance were subtle, but may be an indicator of early symptoms in the development of HD, as suggested by Verny et al.23 MMSE scores are also reported to be correlated with decreased caudate volume, which may be observed from prediagnostic stages of HD.29
Furthermore, sustained attention tasks, such as Stroop test tasks, as well as information processing speed may show alterations in presymptomatic stages of the disease, manifesting before the onset of motor symptoms of HD.29,30 In this study, we observed statistically significant differences in performance in the Stroop-B task (colour naming); although this task involves a degree of automaticity, it requires higher levels of attention than the Stroop-A task. It is reported that automatic responses, which are reflected in performance in the reading condition, deteriorate more frequently in advanced stages of HD.23,29
We identified statistically significant differences between groups and correlations with disease burden in the Stroop test and SDMT; these tests are widely used in the assessment of patients with HD and are highly sensitive.29,31 In particular, SDMT score has been identified as a clinical cognitive marker to assess presymptomatic progression of HD. However, these test include tasks influenced by more than one cognitive process: automaticity and attention in the Stroop test; and attention, visual tracking, and visuomotor coordination in the SDMT. These tests are specifically related to information processing speed, inhibition, attention, and integration of information.
With regard to the verbal fluency test, we only observed statistically significant differences in the phonological fluency task (letter F); these may be related to variations in the cognitive activity demanded by each verbal fluency task. Most research with HD mutation carriers does not report the category or letter used in the different verbal fluency tasks,24,32,33 which make it difficult to confirm whether divergent results in the different studies are explained by the type of test used. It is known that, as the available time to complete the task is limited, information processing speed is an important factor in adequate performance. Therefore, it is likely that verbal fluency deficits will be more evident in tasks with greater lexical requirements, and in subjects with impaired information processing speed.32
Regarding this point, we should consider the possibility that verbal fluency deficits are likely a reflection of the alteration in information processing speed observed in carriers.
According to previous research,29,34,35 the cognitive alterations observed in HD mutation carriers are probably a consequence of striatal and frontostriatal circuit dysfunction, as a result of the loss of striatal projections to the cerebral cortex. Cognitive changes may be linked to biological markers detectable before the onset of motor symptoms or functional impairment. Thus, visuomotor and psychomotor changes may manifest in advanced stages of the disease, whereas alterations in executive performance are observed closer to clinical diagnosis. According to Paulsen et al.,36 cognitive symptoms may be detected approximately 15 years before the onset of motor symptoms and present an insidious progression, which accelerates nearer the diagnosis.
This gives rise to a need to assess cognitive function in early stages; according to Stout et al.,37 neurocognitive tests constitute robust clinical indicators of the disease process before criteria for motor diagnosis of HD are met. Therefore, it has been proposed that neurocognitive performance should be assessed using a comprehensive battery of cognitive tasks designed to maximise the sensitivity for detecting alterations in frontostriatal neuronal circuits. Measuring executive function in individuals at risk of developing HD adds information beyond differences in motor control, and will probably have utilitarian value in clinical settings.38
Lastly, according to the molecular testing results obtained in our study, the mean number of CAG triplet repeats was 43.95 in the carrier group; this is considered a complete penetrance phenotype according to the classification proposed by Tabrizi et al.4 An association is reported between the variable estimated time to onset of the manifest stage of HD and performance in cognitive tests; ie, those who are closer to clinical onset present poorer performance than those with longer time to onset; this is corroborated by our findings. “Mild cognitive impairment” has been observed in up to half of carriers who are close to motor diagnosis.39
ConclusionsHD mutation carriers who are close to symptom onset presented poorer performance than the group of carriers who are far from symptom onset, particularly in neuropsychological tests assessing information processing speed and attention. This confirms that cognitive function, and specifically prefrontal function, is the earliest affected and manifests years before motor diagnosis of HD.
Both cognitive and motor tests that assess the performance of an individual at risk of developing HD may be useful in identifying the onset of several symptoms of HD characterising its severity, in addition to facilitating compensation and stimulation strategies to improve functional capacity and quality of life.
Conflicts of interestThe authors have no conflicts of interest to disclose.
