



Neurofibromatosis type 1 (NF1) is a progressive multisystem disorder following an autosomal dominant inheritance pattern that presents with multiple neurological manifestations.
MethodsWe reviewed medical histories of patients with NF1 followed up at our hospital’s paediatric neurology department from May 1990 to 31 December 2018. We collected data on neurological symptoms.
ResultsA total of 128 patients with NF1 were identified. Mean age (SD) at NF1 diagnosis was 4.43 (3.38) years (range, 0.5-14.5 years). There was a slight female predominance (53.1%). Macrocephaly (head circumference over 2 SDs above average for age) was present in 37.5% of cases. Attention-deficit/hyperactivity disorder was recorded in 28.9% of patients (37): combined type in 20 patients, predominantly inattentive in 15, and predominantly impulsive/hyperactive in 2. Other manifestations included headache (18.6%), cognitive impairment (7.8%), motor deficit (6.2%), and epilepsy (4.68%). Brain MRI was performed in 85 patients, revealing T2-weighted hyperintensities in the basal ganglia and/or cerebellum in 60 patients (70.5%), Chiari malformation type 1 in 4 cases, and arachnoid cysts in 3. Optic nerve gliomas were identified by MRI in 22 patients (25.8%). Other MRI findings included plexiform neurofibromas (9.3%) and central nervous system gliomas (3.1%).
ConclusionsThe neurological manifestations identified in our sample are consistent with those reported in the literature. Effective transfer strategies from paediatric neurology departments and subsequent clinical follow-up by adult neurology departments are needed to prevent loss to follow-up in adulthood.
La Neurofibromatosis tipo 1 (NF1) es un desorden progresivo multisistémico de herencia autosómica dominante que presenta numerosas manifestaciones neurológicas.
MétodosRevisión de historias clínicas de pacientes afectos de NF1 controlados en una Unidad de Neuropediatría de mayo de 1990 a 31 de diciembre de 2018 y sus manifestaciones neurológicas asociadas.
ResultadosSe revisaron 128 pacientes afectos de NF1. Edad media al diagnóstico de NF1, 4,43 años ± 3,38 SDS (Rango 6 meses - 14,5 años) con discreto predominio femenino (53,1%). Se asocia macrocefalia (PC > 2SDS) en el 37,5% de los casos. TDAH en el 28,9% de los casos (37), subtipo combinado 20, inatento 15 casos y predominantemente hiperactivo 2 casos. Otras manifestaciones incluyen; cefalea (18,7%), déficit cognitivo (7,8%), afectación motora (6,2%) y epilepsia (4,68%). Se realizó RM cerebral a 85 pacientes, mostrando 60 (70,5%) hiperseñales en T2 en ganglios basales y/o cerebelo, junto con otras alteraciones como Chiari I (4 casos) y quistes aracnoideos (3 casos). Se identificaron gliomas de nervio óptico en 22 casos (25,8%). Otros hallazgos diagnosticados por RM incluyen neurofibromas plexiformes (9.3%) y otros gliomas localizados en sistema nervioso central (3,1%).
ConclusionesLas manifestaciones neurológicas encontradas concuerdan con lo recogido en la literatura. El seguimiento de estos pacientes se pierde en la edad adulta, siendo necesario establecer adecuadas estrategias de transferencia y posterior seguimiento de pacientes a los servicios de adultos.
Neurofibromatosis type 1 (NF1) is a multisystemic progressive disease presenting an autosomal dominant inheritance pattern and a high rate of spontaneous mutations (50%). Prevalence is estimated at 1 case per 2500 to 3000 population.1
NF1 is caused by mutations in the neurofibromin 1 (NF1) gene, located on chromosome 17q11.2. The NF1 gene product, neurofibromin, is ubiquitously expressed, and complications may therefore affect practically any part of the body at any age, with the appearance and growth of nervous system tumours, together with such other anomalies as skin or bone alterations, being particularly common. Diagnosis was previously based on clinical criteria, but mutations are now identified in more than 95% of cases. NF1 presents great clinical variability and little genotype-phenotype correlation.1
Neurological complications represent one of the most frequent findings.2 Learning and language difficulties, together with inattention, hyperactivity, or impulsivity, are recorded in 30% to 60% of cases.3 Other neurological complications include epilepsy, generally secondary to structural lesions, migraine-like headache, brain glioma, intracranial malformations, aneurysms, and moyamoya syndrome, among others.2
Magnetic resonance imaging (MRI) is the technique of choice for identifying such alterations as optic nerve gliomas, brain tumours, and malformations.4 A high percentage of patients showed T2 hyperintensities in the optic tract, cerebellum, brainstem, thalamus, globus pallidus, and internal capsule; these findings, known as unidentified bright objects, are of uncertain clinical significance and tend to disappear with age.4
We reviewed cases of patients with NF1 attended since May 1990 at the paediatric neurology department, and the associated neurological manifestations.
MethodsOur paediatric neurology department uses clinical protocols, information sheets, and a database of patients assessed since May 1990.5 Since August 2012, we provide an information sheet on NF1. This document explains the condition and the diagnostic tests and periodic monitoring that patients must undergo in accordance with our protocol, which is based on clinical practice guidelines. Follow-up includes an annual or biannual clinical assessment of children younger than 2 years, an annual ophthalmological examination up to the age of 8 years and biennial examination up to the age of 18, annual dermatological examination, comprehensive physical examination to detect bone alterations, annual blood pressure measurement (particularly in adolescents), and monitoring of learning difficulties, among others (Appendix A, Supplementary data).6,7
We performed an observational, descriptive, retrospective study, reviewing the clinical histories of patients with NF1 recorded in the database of the paediatric neurology unit between May 1990 and 31 December 2018, and the neurological manifestations identified.
This study was approved by the clinical research ethics committee of the region of Aragón (file number PI16/092).
ResultsBetween May 1990 and 31 December 2018, our paediatric neurology database included 136 patients with NF1, out of a total of 21 789 patients. We reviewed the clinical histories of 128 patients with NF1, as those of the remaining 8 patients were not available in the hospital’s records due to a long-term lack of follow-up. Ninety-seven patients presented genetic diagnosis of NF1 and 31 were diagnosed according to clinical criteria: these were old cases who did not undergo genetic testing or in whom the techniques used could not identify all mutations.
Mean age (standard deviation) at diagnosis of NF1 was 4.43 (3.38) years (range, 6 months-14.5 years); the sample included 60 boys (46.9%) and 68 girls (53.1%). Mean follow-up time was 6.92 (4.90) years (range, 1 month-20.9 years). Mean age at the last consultation was 10.99 (5.35) years (range, 1-36.5 years; one patient, lost to follow-up from the age of 20 years, consulted at the age of 36 years because his 5-month-old son presented café-au-lait macules).8
Fifty patients are currently under follow-up, 34 girls (68%) and 16 boys (32%), with a mean age of 9.27 (4.16) years (range, 1.5-17.5 years).
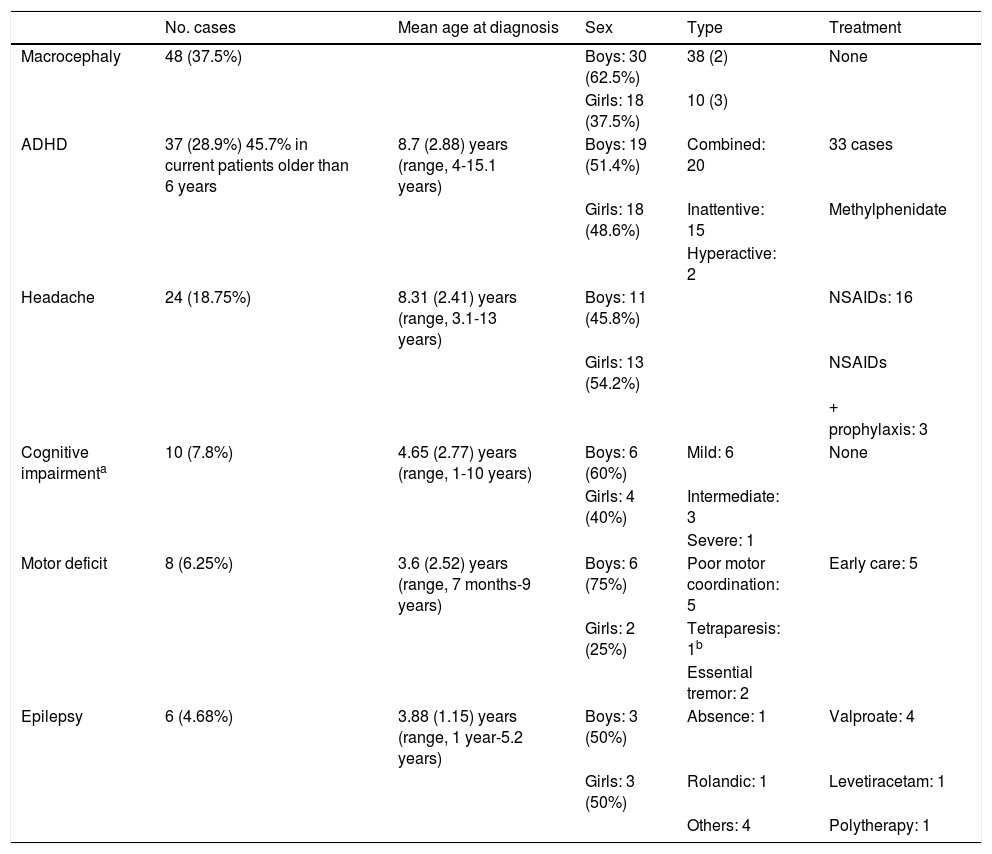
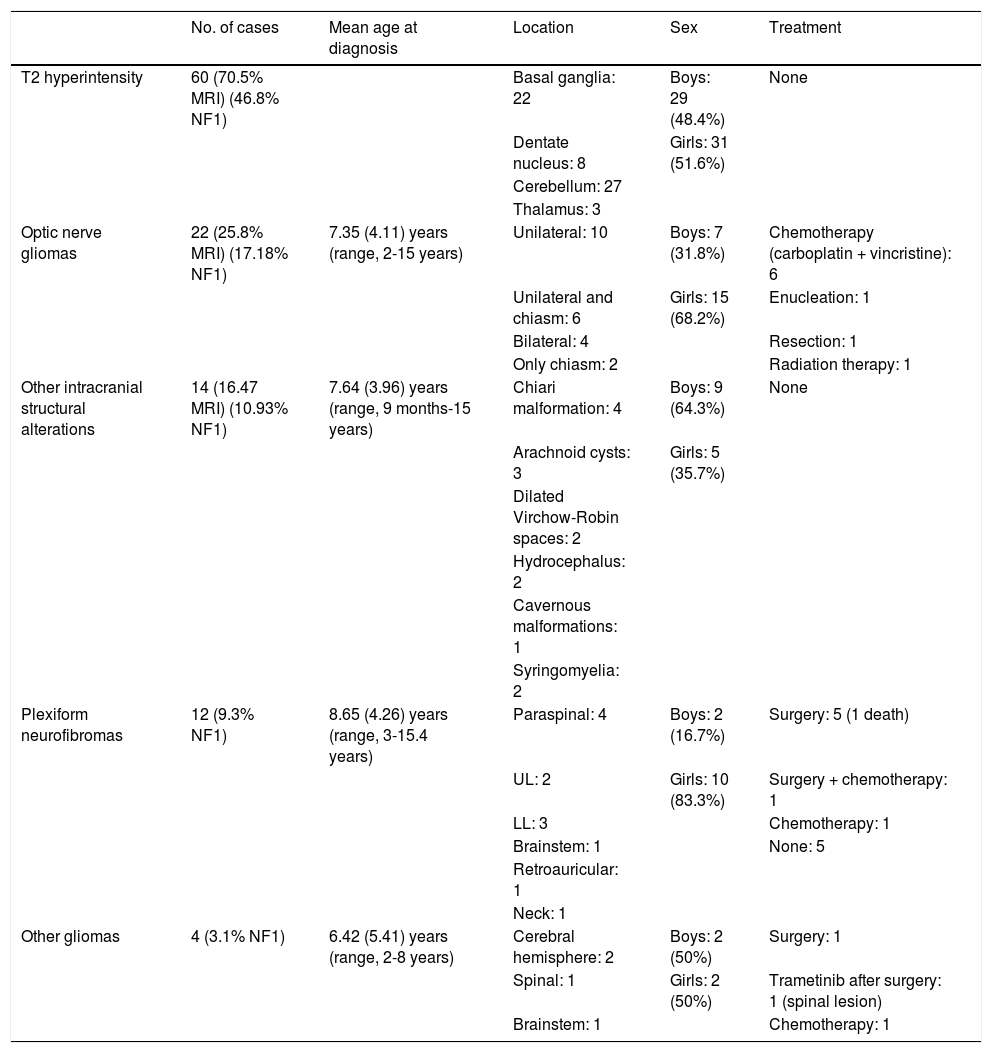
Tables 1 and 2 include the neurological manifestations, MRI findings, and treatments used.
Neurological manifestations in our sample of patients with neurofibromatosis type 1.
| No. cases | Mean age at diagnosis | Sex | Type | Treatment | |
|---|---|---|---|---|---|
| Macrocephaly | 48 (37.5%) | Boys: 30 (62.5%) | 38 (2) | None | |
| Girls: 18 (37.5%) | 10 (3) | ||||
| ADHD | 37 (28.9%) 45.7% in current patients older than 6 years | 8.7 (2.88) years (range, 4-15.1 years) | Boys: 19 (51.4%) | Combined: 20 | 33 cases |
| Girls: 18 (48.6%) | Inattentive: 15 | Methylphenidate | |||
| Hyperactive: 2 | |||||
| Headache | 24 (18.75%) | 8.31 (2.41) years (range, 3.1-13 years) | Boys: 11 (45.8%) | NSAIDs: 16 | |
| Girls: 13 (54.2%) | NSAIDs | ||||
| + | |||||
| prophylaxis: 3 | |||||
| Cognitive impairmenta | 10 (7.8%) | 4.65 (2.77) years (range, 1-10 years) | Boys: 6 (60%) | Mild: 6 | None |
| Girls: 4 (40%) | Intermediate: 3 | ||||
| Severe: 1 | |||||
| Motor deficit | 8 (6.25%) | 3.6 (2.52) years (range, 7 months-9 years) | Boys: 6 (75%) | Poor motor coordination: 5 | Early care: 5 |
| Girls: 2 (25%) | Tetraparesis: 1b | ||||
| Essential tremor: 2 | |||||
| Epilepsy | 6 (4.68%) | 3.88 (1.15) years (range, 1 year-5.2 years) | Boys: 3 (50%) | Absence: 1 | Valproate: 4 |
| Girls: 3 (50%) | Rolandic: 1 | Levetiracetam: 1 | |||
| Others: 4 | Polytherapy: 1 |
ADHD: attention deficit/hyperactivity disorder; NSAIDs: non-steroidal anti-inflammatory drugs.
Structural alterations in magnetic resonance imaging.
| No. of cases | Mean age at diagnosis | Location | Sex | Treatment | |
|---|---|---|---|---|---|
| T2 hyperintensity | 60 (70.5% MRI) (46.8% NF1) | Basal ganglia: 22 | Boys: 29 (48.4%) | None | |
| Dentate nucleus: 8 | Girls: 31 (51.6%) | ||||
| Cerebellum: 27 | |||||
| Thalamus: 3 | |||||
| Optic nerve gliomas | 22 (25.8% MRI) (17.18% NF1) | 7.35 (4.11) years (range, 2-15 years) | Unilateral: 10 | Boys: 7 (31.8%) | Chemotherapy (carboplatin + vincristine): 6 |
| Unilateral and chiasm: 6 | Girls: 15 (68.2%) | Enucleation: 1 | |||
| Bilateral: 4 | Resection: 1 | ||||
| Only chiasm: 2 | Radiation therapy: 1 | ||||
| Other intracranial structural alterations | 14 (16.47 MRI) (10.93% NF1) | 7.64 (3.96) years (range, 9 months-15 years) | Chiari malformation: 4 | Boys: 9 (64.3%) | None |
| Arachnoid cysts: 3 | Girls: 5 (35.7%) | ||||
| Dilated Virchow-Robin spaces: 2 | |||||
| Hydrocephalus: 2 | |||||
| Cavernous malformations: 1 | |||||
| Syringomyelia: 2 | |||||
| Plexiform neurofibromas | 12 (9.3% NF1) | 8.65 (4.26) years (range, 3-15.4 years) | Paraspinal: 4 | Boys: 2 (16.7%) | Surgery: 5 (1 death) |
| UL: 2 | Girls: 10 (83.3%) | Surgery + chemotherapy: 1 | |||
| LL: 3 | Chemotherapy: 1 | ||||
| Brainstem: 1 | None: 5 | ||||
| Retroauricular: 1 | |||||
| Neck: 1 | |||||
| Other gliomas | 4 (3.1% NF1) | 6.42 (5.41) years (range, 2-8 years) | Cerebral hemisphere: 2 | Boys: 2 (50%) | Surgery: 1 |
| Spinal: 1 | Girls: 2 (50%) | Trametinib after surgery: 1 (spinal lesion) | |||
| Brainstem: 1 | Chemotherapy: 1 |
LL: lower limb; MRI: magnetic resonance imaging; NF1: neurofibromatosis type 1; UL: upper limb.
Macrocephaly was the most frequent finding, predominantly occurring in boys.
Headache was associated with structural alterations in 11 cases: 2 cases of Chiari malformations, 2 cases of syringomyelia, one case of dilated Virchow-Robin spaces, 4 cases of optic nerve gliomas, and 2 cases of brain gliomas.
Intellectual disability was observed in 10 patients, and was generally classed as mild, according to the Wechsler Intelligence Scale (Wechsler Preschool and Primary Scale of Intelligence-III and Wechsler Intelligence Scale for Children; scores of 55-70 were considered to indicate mild intellectual disability, 40-55 moderate, and 25-40 severe). Five patients presented some type of associated epilepsy.
Brain MRI was performed in 85 patients (66.4%). The most frequent findings were hyperintensities on T2-weighted sequences, optic nerve gliomas, and intracranial structural alterations. Other MRI findings were plexiform neurofibromas and gliomas in other locations (Table 2).
Optic nerve gliomas were more common among girls (68.2%), and were identified by MRI as an incidental finding in 13 cases and after indication of the study by the ophthalmology department due to such findings as papilloedema (4 cases) and papillary atrophy (5 cases). Four patients presented symptoms before the ophthalmological examination, 3 of whom also presented proptosis together with loss of visual acuity; one patient presented isolated visual impairment. Nine patients received treatment, with 6 receiving combination chemotherapy with carboplatin and vincristine. One patient underwent enucleation of the left eye in 2015 due to significant tumour extension with papillary involvement, despite chemotherapy treatment. Another patient underwent excision of a retrobulbar tumour with sequelae of amaurosis fugax in the right eye at the age of 4 years (1994), and another started corticotherapy followed by radiotherapy in 2009, with partial tumour regression. The remaining patients did not require treatment, and presented no changes in the optic nerve glioma.
One patient, who was lost to follow up from a paediatric age and presented right optic nerve glioma and left internal capsule glioma, consulted at the age of 36.5 years after his 5-month-old son was referred due to café-au-lait macules. An MRI scan requested by his primary care physician in 2012 (after 13 years without follow-up) revealed total regression of the right optic nerve glioma and decreased size of the left internal capsule glioma.8
Most of the other structural alterations detected were incidental findings, and were mainly asymptomatic. We observed ventricular dilation in 2 boys, which was asymptomatic in both cases. In the first case, hydrocephalus was believed to be caused by a left parasagittal T2-hyperintense lesion, which did not cause obstruction or midline shift; in the second case, hydrocephalus was attributed to the presence of multiple hyperintensities causing mild stenosis of the aqueduct of Sylvius. Neither patient required treatment for hydrocephalus after 2.5 and 13 years of follow-up, respectively.
The case of associated cavernomatosis was due to the mutation responsible for multiple familial cerebral cavernomatosis, p.E166X (c.496G>T), located in exon 8 of the PDCD10 gene (CCM3), inherited from the father. The mutation responsible for NF1 had been inherited from the mother.
Plexiform neurofibromas were more frequent in girls (83.3%), with presence of a mass in the affected area being the most frequent manifestation. Three patients presented pain combined with other manifestations: one case of dorsal kyphosis due to presence of dorsal plexiform neurofibroma and 2 cases of gait claudication and impairment due to presence of paraspinal plexiform neurofibroma in the lumbar region. One patient died at the age of 12 years due to exponential growth of a plexiform neurofibroma located in the mediastinum, which recurred despite several surgical resections.
Presence of other gliomas in the brain or other locations was detected incidentally in MRI studies. Due to their size and location, 2 asymptomatic cases were treated with surgical resection of the brainstem glioma and compassionate use of trametinib (a MEK protein inhibitor) for the spinal lesion, as complete resection was not possible. In the case of hemispheric brain gliomas, one case of hypothalamic glioma was treated with chemotherapy due to clinical symptoms of diencephalic syndrome, whereas a watchful waiting approach was adopted in the other case as no growth was observed after 9 years of follow-up.
DiscussionMacrocephaly, defined as a head circumference more than 2 standard deviations greater than average, is the most frequent finding in our sample: 37.5% of our patients displayed this sign, although this is far lower than the rates of 50% to 75% of cases reported by other authors.9–11 Some authors consider macrocephaly to be due to the expansion of some brain structures, mainly the hemispheres, which would in turn cause megalencephaly.12 This growth seems to influence and be related to learning disorders, attention deficit,13 and growth of optic nerve tumours.14
Diagnosis of attention-deficit/hyperactivity disorder (ADHD) was established in approximately 30% of our patients. ADHD affects 45.7% of school-age children currently under follow-up; these figures have increased in recent years. Studies in the literature report ADHD in 38% to 58.33% of patients with NF1.3,15 Diagnosis of ADHD in our sample was established at a mean age of 8.7 (2.88) years, with an age range of 4 to 15 years, similar to the ages reported in the literature: mean of 10.7 (2.2) years, and a range of 4 to 15 years.16 The combined clinical subtype (attention deficit and hyperactivity) is the most frequent (approximately 50%),17 as observed in our sample, although some authors disagree, reporting higher prevalence of the inattentive subtype.18 Treatment with methylphenidate is considered effective in patients with NF1; 33 (89.1%) patients in our sample received this treatment.18,19
Headache is relatively frequent in these patients, although our results (18.75%) contrast with those reported by other series, with up to 50% to 60% of patients presenting headache.20 In these patients, it is important to rule out the presence of other complications as the cause of headache, as was the case in 11 of our patients.20
Intellectual disability is more prevalent in children with NF1 than in the general population; approximately 6% to 7% present an intelligence quotient below 70.21 These rates are similar to that obtained in our study (7.8%), with intellectual disability being mild in most cases. Some authors have associated intellectual disability with the presence of epilepsy in patients with NF122; half of the patients with intellectual disability (5 cases) presented some type of epilepsy.
Seizures affect approximately 6.5% to 9.5% of patients with NF1,21 a rate somewhat higher than that observed in our sample (4.68%). Onset occurs during childhood, as in our series (mean age at diagnosis, 3.88 years [1.15]), or adolescence, although seizures may manifest at any age.23 Focal seizures are most frequent, although patients may also present well-defined symptoms such as absence seizures or West syndrome, among others.24 Patients generally respond adequately to antiepileptic treatment, although some refractory cases may require surgery22,25; in our study, only one patient required combination treatment with several drugs.
MRI is the technique of choice for diagnosing intracranial structural alterations. Indication of MRI is controversial: some authors believe that it should be performed at the time of diagnosis as it may provide diagnostic data, whereas others consider that MRI must be performed in the event of signs suggestive of intracranial anomalies.26 Different clinical practice guidelines currently recommend that MRI should not be performed routinely, but indicate the technique in the event of symptoms suggestive of complications or after detection of complete deletion of the NF1 gene, which is frequently associated with structural brain anomalies.4 In our centre, MRI is performed in patients with clinical symptoms suggestive of some intracranial and/or spinal lesion.
One of the main findings in children with NF1 are hyperintensities on T2-weighted brain MRI sequences, mainly involving the basal ganglia and the cerebellum; these hyperintensities were detected in 70.5% of the MRI scans performed in our sample, which is consistent with the figures reported in other series (50%-90%).4 They coincide with intramyelinic vacuolation,27 which is more frequent in early ages and is of uncertain clinical significance.4 Some authors associate these findings with the onset of learning disorders and cognitive impairment,28 although no conclusive data are available.29
Optic nerve gliomas are the most frequent central nervous system tumours in patients with NF1, with a prevalence rate of 15% to 25%,30 which is comparable with our findings (17.18%). These tumours are generally asymptomatic, and are more frequent in children younger than 6 years (mean age of 7.35 [4.11] at diagnosis in our series); from the age of 6 to 7 years, slow growth and even a spontaneous regression may be observed.8,31 A watchful waiting approach with periodic assessment is followed in asymptomatic cases. In symptomatic cases, chemotherapy may be necessary and is generally the treatment of first choice, with a combination of such drugs as carboplatin/cisplatin, vincristine, or a MEK inhibitor. Radiotherapy should be avoided, and should only be used after exhausting the second and third line of treatment with chemotherapy or MEK inhibitors.
Surgery is reserved for those cases with such severe symptoms as proptosis, significant aesthetic impact, and vision loss.30,32
Patients with optic pathway glioma present higher probability (20%) of developing low-grade glioma, generally pilocytic or pilomyxoid astrocytoma, in other locations of the central nervous system.30 They represent 2% to 3% of the patients with NF1, a similar rate to that observed in our study (3.1%).
Other intracranial structural alterations are observed in approximately 5% of patients with NF1,33,34 although this percentage was slightly higher in our sample (10.9%).
Type I Chiari malformation has been detected in 5% of the population with NF1, and is generally asymptomatic, although it may occasionally be associated with such other manifestations as headache and neck pain.35
Arachnoid cysts may appear in patients with NF1, although their prevalence is difficult to determine; although generally asymptomatic, they may cause symptoms of intracranial hypertension, seizures, and visual alterations due to compression in the event of cyst rupture or growth.36,37
Although presence of cavernomas has not been associated with NF1, some authors have attempted to establish a relationship between these entities in recent years.34,38 In our patient presenting both conditions, the association is clearly coincidental, with one mutation inherited from the father (cavernomatosis) and the other from the mother (NF1).
Hydrocephalus was rare in our sample (1.5%), but this rate is similar to those reported by other authors, who estimate an incidence rate between 1% and 5%.39 It is generally caused by aqueductal stenosis associated with tumours, as was the case in one of our patients, or cranial base pathology.40
Plexiform neurofibromas are more frequent (20%) than the rate reported in our study (9.3%). They may go undetected and be asymptomatic, mainly in paediatric patients, as their growth is more marked from adolescence, which may delay diagnosis2; in our case, the mean age at diagnosis was approximately 8.5 years. In symptomatic cases, plexiform neurofibromas may be incapacitating and require surgical treatment,41 as was the case for 6 of our patients, and/or new therapies, including selumetinib, a selective inhibitor of MEK 1 and 2 kinase that may induce tumour regression and has been shown to decrease the size of plexiform neurofibroma in patients with NF1 in a clinical trial.42
Our patients were lost to follow-up, as there is no specific consultation for adults with NF1. Chronic diseases, and especially hereditary and rare diseases, require lifelong monitoring. Adequate referral strategies and subsequent follow-up of patients at adult consultations should be implemented. Properly informing patients and monitoring the associated risks is a huge responsibility, as are genetic counselling and prenatal diagnosis. Long-term follow-up and adequate communication between paediatric and adult specialists are essential tools for better management of our patients, who may benefit from continuous technical, scientific, and social advances, and for our understanding of the natural progression of diseases.
Conflicts of interestThe authors have no conflicts of interest to declare.
The following is Supplementary data to this article:
Please cite this article as: Sánchez Marco SB, López Pisón J, Calvo Escribano C, González Viejo I, Miramar Gallart MD, Samper Villagrasa P. Manifestaciones neurológicas en neurofibromatosis tipo 1. Nuestra experiencia. Neurología. 2022;37:325–333.
