




FOXG1 syndrome is an epileptic-dyskinetic encephalopathy initially described as a variant of Rett syndrome (OMIM #613454). In most cases, this neurodevelopmental disorder is caused by mutations in the MECP2 gene; it exclusively affects women. Rett syndrome is characterised by developmental regression between 6 and 18 months of age, stereotypic hand movements, microcephaly, seizures, and intellectual disability. The condition presents a broad phenotypic spectrum and genetic heterogeneity: it has been associated with microdeletions and translocations in various genes, including CDKL5 (also known as STK9), NTNG1, and more recently FOXG1. FOXG1, located on chromosome 14q12, plays a significant role in brain development,1 regulating early embryonic development of the telencephalon in the fetus; it is also expressed in adulthood.2 We present the case of a patient with a heterozygous mutation in FOXG1, which supports the notion that FOXG1 syndrome is a distinct entity rather than a variant of Rett syndrome.
Our patient, a 3-year-old boy from Colombia, was born to consanguineous parents following an uneventful second pregnancy. He was born in week 37 of pregnancy by caesarean delivery, with a birth weight of 2680g (10th-50th percentile), length of 47cm (10th-50th percentile), and a head circumference of 35.5cm (50th-90th percentile). No perinatal complications were recorded; an evaluation performed at 3 months by the paediatric neurology department revealed a bulging anterior fontanelle and slightly increased muscle tone. An axial CT scan of the head with 3D reconstruction revealed open opercula, enlarged zygomatic bones, no craniosynostosis, microcephaly, flat occiput, and head circumference 2 standard deviations (SD) below the mean. The patient was further assessed by the paediatric ophthalmology and clinical genetics departments due to psychomotor retardation and gastro-oesophageal reflux disease. The ophthalmological examination revealed partial binocular fixation, repetitive head movements, and abnormal visual behaviour for his age, associated with global developmental delay.
At 3 years of age, the patient had a weight of 8.7kg (−2.25 SD), a height of 82cm (−0.53 SD), a low weight-to-height ratio (−2.75 SD), a BMI of 12.9kg/m2 (−2.91 SD), a head circumference of 40cm (−2 SD), microcephaly, flat occiput, prominent metopic ridge, prominent forehead, convergent strabismus, low nasal bridge, smooth philtrum, anteverted ears, tongue protrusion, and shawl scrotum. A metabolic study revealed no alterations and tests for mucopolysaccharidosis yielded negative results. G-banding revealed a normal chromosome complement (46,XY) and findings from microarray-based comparative genomic hybridisation were normal with regions of homozygosity. Exome sequencing of the patient and his parents was performed with the Illumina TruSight One sequencing panel kit and the Next Seq 500 system. An analysis of the genes related to our patient's clinical diagnosis revealed a heterozygous mutation in FOXG1 (NM_005249.4): c.1107:1108insG, p.Glu371GlyfsTer84, in which the insertion of a guanine results in a premature stop codon, causing a truncated protein. This variant has not previously been reported. However, the literature describes mutations affecting the transcription of the same amino acid (c.460dupG, p.e154GfsX301), which are reported to be pathogenic as they result in the loss of 3 protein-binding domains.3 Presence of the candidate variant was confirmed with Sanger sequencing; DNA sequencing of the parents revealed that this was a de novo mutation. In summary, our patient displayed poor growth, postnatal microcephaly, poor visual contact and social interaction, convergent strabismus, tongue protrusion, gastro-oesophageal reflux disease, flat feet, neonatal hypotonia, neonatal irritability, delayed motor development, apraxia, stereotypic movement, lack of language, poor sleep pattern, EEG alterations, normal MRI findings, and a de novo mutation. These findings show a genotype-phenotype correlation with atypical Rett syndrome secondary to FOXG1 mutations.
FOXG1 mutations are extremely rare, presenting in 1%-2% of patients with suspected autism spectrum disorders and 1.5% of female patients with severe intellectual disability and microcephaly.4FOXG1 acts as a transcriptional repressor. It is highly expressed in the ventricular side of the neuroepithelium in the telencephalon and in visual structures and testicular tissue,5 promoting the proliferation of progenitor cells and early suppression of neurogenesis and neuronal differentiation.6 This is achieved through the recruitment of transcriptional repressor proteins Groucho and JARID1B, which participate in the methylation of histone 3 at lysine 4 residues, resulting in chromatin silencing.7 Molecular analyses have shown that FOXG1 shares metabolic pathways with MECP2 during neuronal development; furthermore, the 2 genes show similar expression profiles in postnatal cortical and subnuclear tissue.8
FOXG1 is the candidate gene for monogenic 14q12 microdeletion syndrome. Our patient had psychomotor delay, in spite of which he was able to sit up and walk. This illustrates the haploinsufficiency of FOXG1, which causes less severe manifestations; a large proportion of the patients described in the literature reach such developmental milestones as walking (3 out of 8 patients). Some studies conclude that heterozygous FOXG1 variants do not necessarily cause brain malformations,9 as exemplified by our case.
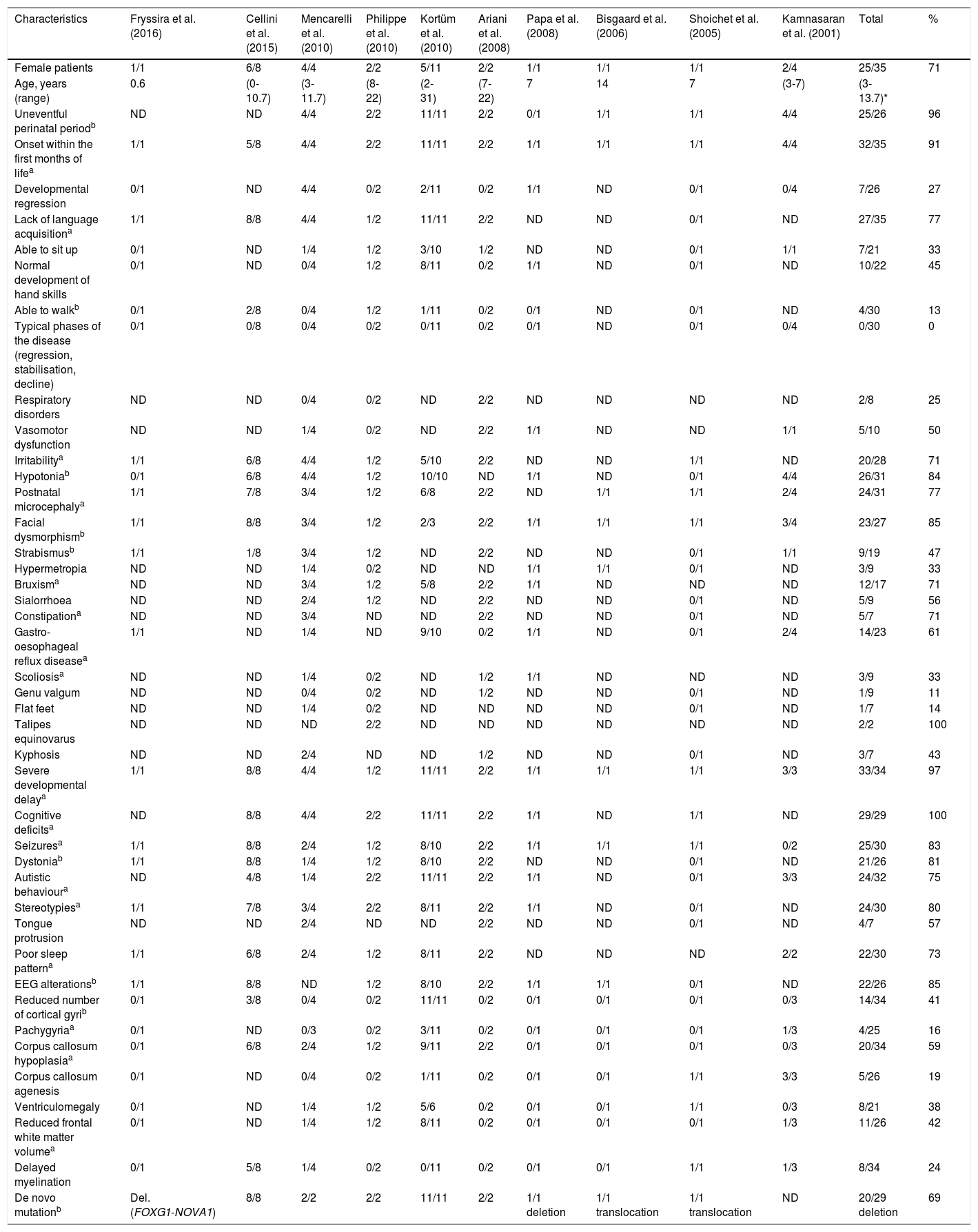
Atypical Rett syndrome has been linked to mutations in several genes, including CDKL5 (also known as STK9), NTNG1, and FOXG1. Clinical manifestations in FOXG1 mutation carriers, however, are considerably different from those observed in patients with Rett syndrome. This explains why some authors suggest that FOXG1 syndrome is clinically and genotypically distinct from Rett syndrome and presents such distinctive clinical characteristics as hyperkinetic dyskinesia.10 We analysed the published cases of 35 patients with de novo point mutations, deletions, and translocations in FOXG1, with clinical phenotypes depending on a wide genotypic variability. FOXG1 syndrome affects both sexes (69% females). A large proportion of these patients do not show developmental regression or the typical stages of Rett syndrome; on the contrary, the associated alterations appear in the neonatal period. Table 1 summarises the characteristics of FOXG1 syndrome described by Kortüm et al.10; we have added several other characteristics appearing in over 75% of the published cases, expanding and better defining the phenotype of the syndrome.
Characteristics of the cases of FOXG1 syndrome reported in the literature.
| Characteristics | Fryssira et al. (2016) | Cellini et al. (2015) | Mencarelli et al. (2010) | Philippe et al. (2010) | Kortüm et al. (2010) | Ariani et al. (2008) | Papa et al. (2008) | Bisgaard et al. (2006) | Shoichet et al. (2005) | Kamnasaran et al. (2001) | Total | % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Female patients | 1/1 | 6/8 | 4/4 | 2/2 | 5/11 | 2/2 | 1/1 | 1/1 | 1/1 | 2/4 | 25/35 | 71 |
| Age, years (range) | 0.6 | (0-10.7) | (3-11.7) | (8-22) | (2-31) | (7-22) | 7 | 14 | 7 | (3-7) | (3-13.7)* | |
| Uneventful perinatal periodb | ND | ND | 4/4 | 2/2 | 11/11 | 2/2 | 0/1 | 1/1 | 1/1 | 4/4 | 25/26 | 96 |
| Onset within the first months of lifea | 1/1 | 5/8 | 4/4 | 2/2 | 11/11 | 2/2 | 1/1 | 1/1 | 1/1 | 4/4 | 32/35 | 91 |
| Developmental regression | 0/1 | ND | 4/4 | 0/2 | 2/11 | 0/2 | 1/1 | ND | 0/1 | 0/4 | 7/26 | 27 |
| Lack of language acquisitiona | 1/1 | 8/8 | 4/4 | 1/2 | 11/11 | 2/2 | ND | ND | 0/1 | ND | 27/35 | 77 |
| Able to sit up | 0/1 | ND | 1/4 | 1/2 | 3/10 | 1/2 | ND | ND | 0/1 | 1/1 | 7/21 | 33 |
| Normal development of hand skills | 0/1 | ND | 0/4 | 1/2 | 8/11 | 0/2 | 1/1 | ND | 0/1 | ND | 10/22 | 45 |
| Able to walkb | 0/1 | 2/8 | 0/4 | 1/2 | 1/11 | 0/2 | 0/1 | ND | 0/1 | ND | 4/30 | 13 |
| Typical phases of the disease (regression, stabilisation, decline) | 0/1 | 0/8 | 0/4 | 0/2 | 0/11 | 0/2 | 0/1 | ND | 0/1 | 0/4 | 0/30 | 0 |
| Respiratory disorders | ND | ND | 0/4 | 0/2 | ND | 2/2 | ND | ND | ND | ND | 2/8 | 25 |
| Vasomotor dysfunction | ND | ND | 1/4 | 0/2 | ND | 2/2 | 1/1 | ND | ND | 1/1 | 5/10 | 50 |
| Irritabilitya | 1/1 | 6/8 | 4/4 | 1/2 | 5/10 | 2/2 | ND | ND | 1/1 | ND | 20/28 | 71 |
| Hypotoniab | 0/1 | 6/8 | 4/4 | 1/2 | 10/10 | ND | 1/1 | ND | 0/1 | 4/4 | 26/31 | 84 |
| Postnatal microcephalya | 1/1 | 7/8 | 3/4 | 1/2 | 6/8 | 2/2 | ND | 1/1 | 1/1 | 2/4 | 24/31 | 77 |
| Facial dysmorphismb | 1/1 | 8/8 | 3/4 | 1/2 | 2/3 | 2/2 | 1/1 | 1/1 | 1/1 | 3/4 | 23/27 | 85 |
| Strabismusb | 1/1 | 1/8 | 3/4 | 1/2 | ND | 2/2 | ND | ND | 0/1 | 1/1 | 9/19 | 47 |
| Hypermetropia | ND | ND | 1/4 | 0/2 | ND | ND | 1/1 | 1/1 | 0/1 | ND | 3/9 | 33 |
| Bruxisma | ND | ND | 3/4 | 1/2 | 5/8 | 2/2 | 1/1 | ND | ND | ND | 12/17 | 71 |
| Sialorrhoea | ND | ND | 2/4 | 1/2 | ND | 2/2 | ND | ND | 0/1 | ND | 5/9 | 56 |
| Constipationa | ND | ND | 3/4 | ND | ND | 2/2 | ND | ND | 0/1 | ND | 5/7 | 71 |
| Gastro-oesophageal reflux diseasea | 1/1 | ND | 1/4 | ND | 9/10 | 0/2 | 1/1 | ND | 0/1 | 2/4 | 14/23 | 61 |
| Scoliosisa | ND | ND | 1/4 | 0/2 | ND | 1/2 | 1/1 | ND | ND | ND | 3/9 | 33 |
| Genu valgum | ND | ND | 0/4 | 0/2 | ND | 1/2 | ND | ND | 0/1 | ND | 1/9 | 11 |
| Flat feet | ND | ND | 1/4 | 0/2 | ND | ND | ND | ND | 0/1 | ND | 1/7 | 14 |
| Talipes equinovarus | ND | ND | ND | 2/2 | ND | ND | ND | ND | ND | ND | 2/2 | 100 |
| Kyphosis | ND | ND | 2/4 | ND | ND | 1/2 | ND | ND | 0/1 | ND | 3/7 | 43 |
| Severe developmental delaya | 1/1 | 8/8 | 4/4 | 1/2 | 11/11 | 2/2 | 1/1 | 1/1 | 1/1 | 3/3 | 33/34 | 97 |
| Cognitive deficitsa | ND | 8/8 | 4/4 | 2/2 | 11/11 | 2/2 | 1/1 | ND | 1/1 | ND | 29/29 | 100 |
| Seizuresa | 1/1 | 8/8 | 2/4 | 1/2 | 8/10 | 2/2 | 1/1 | 1/1 | 1/1 | 0/2 | 25/30 | 83 |
| Dystoniab | 1/1 | 8/8 | 1/4 | 1/2 | 8/10 | 2/2 | ND | ND | 0/1 | ND | 21/26 | 81 |
| Autistic behavioura | ND | 4/8 | 1/4 | 2/2 | 11/11 | 2/2 | 1/1 | ND | 0/1 | 3/3 | 24/32 | 75 |
| Stereotypiesa | 1/1 | 7/8 | 3/4 | 2/2 | 8/11 | 2/2 | 1/1 | ND | 0/1 | ND | 24/30 | 80 |
| Tongue protrusion | ND | ND | 2/4 | ND | ND | 2/2 | ND | ND | 0/1 | ND | 4/7 | 57 |
| Poor sleep patterna | 1/1 | 6/8 | 2/4 | 1/2 | 8/11 | 2/2 | ND | ND | ND | 2/2 | 22/30 | 73 |
| EEG alterationsb | 1/1 | 8/8 | ND | 1/2 | 8/10 | 2/2 | 1/1 | 1/1 | 0/1 | ND | 22/26 | 85 |
| Reduced number of cortical gyrib | 0/1 | 3/8 | 0/4 | 0/2 | 11/11 | 0/2 | 0/1 | 0/1 | 0/1 | 0/3 | 14/34 | 41 |
| Pachygyriaa | 0/1 | ND | 0/3 | 0/2 | 3/11 | 0/2 | 0/1 | 0/1 | 0/1 | 1/3 | 4/25 | 16 |
| Corpus callosum hypoplasiaa | 0/1 | 6/8 | 2/4 | 1/2 | 9/11 | 2/2 | 0/1 | 0/1 | 0/1 | 0/3 | 20/34 | 59 |
| Corpus callosum agenesis | 0/1 | ND | 0/4 | 0/2 | 1/11 | 0/2 | 0/1 | 0/1 | 1/1 | 3/3 | 5/26 | 19 |
| Ventriculomegaly | 0/1 | ND | 1/4 | 1/2 | 5/6 | 0/2 | 0/1 | 0/1 | 1/1 | 0/3 | 8/21 | 38 |
| Reduced frontal white matter volumea | 0/1 | ND | 1/4 | 1/2 | 8/11 | 0/2 | 0/1 | 0/1 | 0/1 | 1/3 | 11/26 | 42 |
| Delayed myelination | 0/1 | 5/8 | 1/4 | 0/2 | 0/11 | 0/2 | 0/1 | 0/1 | 1/1 | 1/3 | 8/34 | 24 |
| De novo mutationb | Del. (FOXG1-NOVA1) | 8/8 | 2/2 | 2/2 | 11/11 | 2/2 | 1/1 deletion | 1/1 translocation | 1/1 translocation | ND | 20/29 deletion | 69 |
a Characteristics described by Kortüm et al.
b Characteristics frequently reported in the literature.
* Interquartile range.
Del.: deletion; ND: no data.
FOXG1 syndrome is caused by mutations; as is the case with such chromosomal rearrangements as deletions or translocations in 14q12-q13, the condition causes epileptic-dyskinetic encephalopathy, with a well-defined phenotype.10 FOXG1 syndrome was first described as a variant of Rett syndrome, as both conditions share clinical characteristics; however, they also present significant differences. Table 2 lists the major and minor clinical features of the syndrome, according to the frequency reported in the literature, and the most frequent imaging findings.
Major and minor diagnostic criteria and imaging characteristics of FOXG1 syndrome.
| Major diagnostic criteria |
| 1. De novo mutation (translocation or deletion) |
| 2. Cognitive deficits |
| 3. Uneventful perinatal period, onset within the first months of life |
| 4. Postnatal microcephaly |
| 5. Hypotonia |
| 6. Severe developmental delay, lack of language acquisition |
| 7. EEG alterations |
| Minor diagnostic criteria |
| Irritability, facial dysmorphism (prominent forehead, anteverted ears, hypertelorism), strabismus, bruxism, constipation, gastro-oesophageal reflux disease, seizures, dystonia, poor sleep pattern, stereotypies, autistic behaviour |
| Imaging criteria |
| Pachygyria, reduced number of cortical gyri, corpus callosum hypoplasia, reduced frontal white matter volume |
This study was funded by Icesi University.
Please cite this article as: Candelo E, Caicedo G, Pachajoa H. Ampliando el fenotipo del síndrome FOXG1. Neurología. 2020;35:207–211.
