


We report the case of a newborn (NB), the first son of non-consanguineous parents, diagnosed with intrauterine growth delay by ultrasonography in the sixth month of pregnancy. Delivery occurred at 40 weeks, and the infant had a weight of 2440g (−2.59SD) and a length of 47cm (−2.2SD).1 Blood glucose at 3h was 28mg/dL, and intravenous (4mg/kg/min) and oral glucose was therefore provided. Hyperglycemia greater than 200mg/dL started on the fifth day, which prompted intravenous insulin therapy with a dose up to 3.3IU/kg/h. No ketosis was found. Treatment with subcutaneous insulin was subsequently maintained at a dose of 7IU/kg/day until two and a half months of age, when it was discontinued due to frequent hypoglycemia episodes.
A physical examination of the NB revealed a unique phenotype, elfin facies with low-set ears, flat nasal root, flaring nostrils, macrostomia, acanthosis nigricans in multiple sites, hypertrichosis, pachyderma with no fatty layer in the chest and arms, a small chest, and a very developed scrotum.
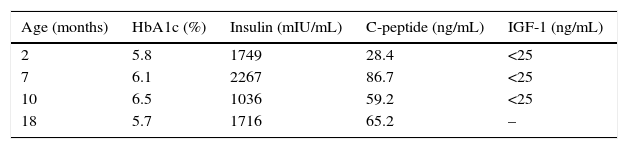
At 3 months of life and with no insulin therapy, the basal insulin level was 1749mIU/mL (Table 1) with negative GAD, IA2, and insulin antibodies. A genetic study revealed two compound heterozygous mutations in the INSR gene, encoding insulin receptor (p.R1026X2/D1179GfsX8), the latter being a mutation not reported in the literature. Both parents were given an oral glucose tolerance test and the results were normal, with a HOMA index of 1.6 in the father and of 2.9 in the mother.
The patient eventually experienced multiple episodes of fasting and late postprandial hypoglycemia. It was therefore decided to start oral feeding every 2h (starting formula, dextrin–maltose, and MCTs) because the family rejected continuous feeding through nasogastric tube. After this, weekly blood glucose measurements lower than 50mg/dL decreased from 20 to less than 1–2, and glycemic control stabilized. Weight and height development, after recovery in the first few months, was −1.8SD for height (target height −1.5SD) and 0SD for weight. Psychic development and learning were adequate, but motor development was delayed due to marked muscle hypotonia. The infant experienced frequent respiratory infections and died at 19 months of age from a serious respiratory tract infection that required admission to the intensive care unit.
This was a case of severe insulin resistance. The genetic causes of insulin resistance include genetic defects in insulin receptor (type A insulin resistance, Rabson–Mendehall syndrome, and leprechaunism), lipodystrophy (Seip–Berardinelli syndrome and Kobberling–Dunnigan syndrome), and immune changes (type B insulin resistance and ataxia-telangiectasia syndrome).3 The reported patient had extreme insulin resistance due to a mutation in the insulin receptor gene in its most severe expression, Donohue syndrome or leprechaunism. This extremely uncommon syndrome was described in 19544 and occurs in approximately one out of every 4,000,000 NBs. The syndrome is associated with a unique elfin phenotype. It is a recessive autosomal disease caused by mutations in the gene that encodes the insulin receptor, located in chromosome 19 (19p13.2).5 This receptor consists of 22 exons5; both the mutations identified in this patient affected the tyrosine kinase domain of the receptor. The first mutation was located in exon 17, with the replacement of arginine by a stop codon at position 1026, while the second mutation (D1179GfsX8) generated a change at the splicing site between exons 19 and 20, resulting in an altered protein from amino acid 1179 that generated a premature stop codon.
The severity of the disease and phenotype is determined by the severity of the mutations, with impaired receptor function being associated with a poorer prognosis and a decreased chance of survival.6 A wide genetic heterogeneity exists, and multiple mutations have been reported.7 Diagnosis is based on clinical signs and symptoms (pre- and postnatal growth delay, soft tissue overgrowth, and the poor development of muscle and adipose tissue, requiring insulin action), biochemical changes including fasting hypoglycemia and postprandial hyperglycemia, extreme hyperinsulinism, and mutations in the insulin receptor gene.
In extreme insulin resistance syndromes, impaired glucose metabolism includes both insulin-resistant diabetes and hypoglycemia, which may precede hyperglycemia. Hypoglycemia may occur both under fasting conditions and in the late postprandial period (3–4h after intake)8 and typically occurs in these patients, particularly after food intake. Under these conditions there is no balance between insulin and glucose, and an overcompensation that causes hypoglycemia sometimes occurs. There is also a marked delay in the hepatic clearance of insulin,3 which may contribute to late hypoglycemic episodes. Marked insulin resistance also contributes to a state of hyperandrogenism which in females causes polycystic ovary disease that may be massive. Histological changes secondary to hyperinsulinism are also seen, including hyperplasia of β cells, breasts, epithelial tissue of skin, renal tubules and bronchi.
All of these, combined with almost complete lymphoid tissue atrophy, contribute to a high susceptibility to infections, particularly in the respiratory tract. A very small chest and muscle hypotonia are factors that aggravate infections. Life expectation is usually very short, and most children die before 2 years of age.9
No curative treatment exists. Diabetes may be treated with insulin or insulin sensitizers, and when hypoglycemia is the predominant clinical symptom, as occurred in our case, very frequent, continuous oral feeding through a gastric or oral tube appears to be effective.10 With this approach, the number of hyperglycemic episodes was greatly reduced in our patient. On the other hand, in patients treated with high doses of recombinant human IGF-1 there is evidence of improvements in metabolic control in the event of diabetes and in growth pattern, although variable results have been reported.11 A multidisciplinary approach to this type of patient is recommended, focusing on optimizing glycemic control, but also on trying to prevent, to whatever extent possible, complications such as respiratory tract infections, which in the end only worsen the prognosis.
FundingThe authors state that they have received no funding for the conduct of this work.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Sánchez-Hernández RM, Martín-Frías M, Castaño L, Lamas A, Barrio R. Síndrome de Donohue. Resistencia extrema a la insulina en el periodo neonatal. Endocrinol Nutr. 2016;63:45–46.
