




Ectopic Cushing's syndrome (ECS) is a rare condition caused by ACTH secretion by extrapituitary tumors. Its low frequency makes it difficult to acquire experience in its management. The aim of this study was to describe patients with ECS seen at the endocrinology department of a tertiary hospital over 15 years.
MethodsThis was a retrospective study of the clinical, biochemical and radiographic data, treatment, and course of patients with ECS seen from 2000 to 2015.
ResultsNine patients (6 of them female) with a mean age of 47 years were included in the study. The clinical syndrome developed in less than 3 months in all cases but one, and most patients also had edema, hyperpigmentation and/or hypokalemia. Mean urinary free cortisol and ACTH levels were 2840μg/24h and 204pg/mL respectively. The ectopic origin was confirmed by a combination of dynamic non-invasive tests and radiographic studies in most cases. The tumor responsible could be identified in 8 cases, and 7 patients had metastatic dissemination. Primary treatment was surgery in one patient, surgery combined with systemic therapy in 3, and chemotherapy in the other 3 patients. Bilateral adrenalectomy was required in 4 patients to control hypercortisolism. After a mean follow-up of 40 months, 3 patients died, 5 were still alive, and one had been lost to follow-up.
ConclusionsOur study confirms that ECS covers a wide spectrum of tumors of different aggressiveness and nature. The ectopic origin of Cushing's syndrome can usually, be suspected and confirmed in most cases without the need for invasive tests. Control of both hypercortisolism and the tumor requires multiple treatment modalities, and multidisciplinary management is recommended.
El síndrome de Cushing ectópico (SCE) es una entidad rara debida a la secreción de ACTH por tumores extrahipofisarios. Su baja frecuencia dificulta la adquisición de experiencia en su manejo. El objetivo de este trabajo es describir a los pacientes con SCE atendidos en el servicio de Endocrinología en un hospital de tercer nivel en un periodo de 15 años.
MétodosSe trata de un estudio retrospectivo de los datos clínicos, bioquímicos y radiológicos, tratamiento recibido, y evolución de los pacientes con SCE atendidos entre los años 2000 y 2015.
ResultadosSe incluyeron 9 pacientes (6 mujeres) con una edad media de 47 años. El síndrome clínico se desarrolló en un tiempo inferior a 3 meses en todos los casos excepto en uno, y la mayoría presentaba edemas, hiperpigmentación y/o hipopotasemia. La media del cortisol libre urinario y de la ACTH fue de 2.840μg/24h y 204pg/ml, respectivamente. El origen ectópico se confirmó por la combinación de pruebas dinámicas no invasivas y estudios radiológicos en la mayoría de los casos. El tumor responsable pudo identificarse en 8 casos y 7 presentaban diseminación metastásica. El tratamiento primario consistió en cirugía en un caso, cirugía más terapia sistémica en 3 y quimioterapia en otros 3. En 4 pacientes fue necesaria la suprarrenalectomía bilateral para controlar el hipercortisolismo. Tras un seguimiento medio de 40 meses, 3 habían fallecido, 5 permanecían vivos y en uno se había perdido el seguimiento.
ConclusionesSe confirma que el SCE abarca un amplio espectro de tumores de diferente agresividad y naturaleza. Habitualmente el origen ectópico del síndrome de Cushing puede sospecharse y confirmarse en la mayoría de los casos sin necesidad de pruebas invasivas. Tanto el control del hipercortisolismo como del tumor requieren múltiples modalidades terapéuticas, siendo recomendable el manejo multidisciplinar.
The secretion of adrenocorticotropic hormone (ACTH) by extrapituitary tumors as the cause of Cushing's syndrome (CS) was described by Liddle in 1962, and accounts for approximately 15% of all cases of endogenous CS.1,2 The ACTH source usually corresponds to neuroendocrine differentiation tumors of highly variable location and aggressivity. In 60% of the cases the tumor is intrathoracic (small cell lung carcinoma or bronchial carcinoid and thymic tumors). The remaining presentations comprise mainly neuroendocrine pancreatic tumors, pheochromocytomas and medullary thyroid carcinomas.3–8
The incidence is similar in both genders, and the mean patient age at presentation is 40–50 years, though cases have also been documented in children and adolescents.9 The clinical presentation covers a broad spectrum from fulminant forms of hypercortisolism in which myopathy, edema, hyperpigmentation and psychiatric alterations predominate, to other forms characterized by a more indolent course with symptoms similar to those typical of Cushing's disease (CD). In the latter case, establishing the differential diagnosis between ectopic CS (ECS) and CD constitutes a genuine challenge.3 Likewise, identification of the ACTH source and the treatment of hypercortisolism often pose considerable difficulties.
The present study describes our experience with the diagnosis, treatment, and long-term prognosis of patients with ECS seen in our center over a 15-year period.
Material and methodsPatientsThe study included patients with ECS seen in the Department of Endocrinology of Hospital Puerta de Hierro (Majadahonda, Madrid, Spain) between 2000 and 2015. Carriers of ECS were defined as patients with ACTH-dependent CS who moreover met some of the following criteria suggesting extrapituitary ACTH secretion: (a) suggestive clinical and biochemical manifestations with evidence of a tumor strongly suspected to be of a neuroendocrine nature; (b) two consistent dynamic test results and evidence of a neuroendocrine tumor; or (c) consistent inferior petrosal sinus catheterization (IPSC) findings.
Biochemical studyIn all cases the biochemical study included the measurement of serum cortisol (SC) at 8:00a.m. on at least two occasions (normal range [NR]: 4.3–22.4μg/dl), basal ACTH (8:00a.m.) (NR: 9–55pg/ml), and 24-h urinary free cortisol (UFC) (NR: 11–71μg/24h). Depending on the diagnostic difficulties encountered, in some cases we also conducted one or more of the following tests: nocturnal SC (23:00p.m.) (NR<7.5μg/dl), a high-dose dexamethasone (DXM) suppression test (DST) (2mg dexamethasone/6h for 2 days), a CRH test, and inferior petrosal sinus catheterization. We defined suppression in DST as a decrease in UFC>90% and/or SC >50% versus the basal levels. The CRH test was considered positive for CD in the case of ACTH elevation >50% and/or SC elevation >20% versus the basal levels. In inferior petrosal sinus catheterization, a central/peripheral ACTH gradient <2 under basal conditions and <3 after CRH was considered indicative of ectopic ACTH secretion.
Serum cortisol and ACTH were measured by chemiluminescence (Advia Centaur XP, Siemens Healthineers, Erlangen, Germany). Twenty-four hour urinary free cortisol was measured by radioimmunoassay (Stratec, Diasource, Louvain-la-Neuve, Belgium).
Imaging studiesComputed tomography (CT) or magnetic resonance imaging (MRI) was performed in all patients. In addition, one or more of the following procedures were used in some cases: scintigraphy with 111In DTPA-octreotide (octreoscan), scintigraphy with 123I-metaiodobenzylguanidine (MIBG), positron emission tomography (PET) with fluorodeoxyglucose (18FDG), and PET with fluorodopa (18F-DOPA).
Other explorations performed to identify the source of ACTH or confirm a suspected neuroendocrine tumor were: (a) the quantification of tumor markers: chromogranin A, gastrin, insulin, glucagon, vasoactive intestinal peptide (VIP), somatostatin, 5-hydroxyindoleacetic acid, carcinoembryonic antigen (CEA), calcitonin, and fractionated metanephrines in 24-h urine; (b) gastrointestinal and/or bronchial endoscopic studies; and (c) histological sampling.
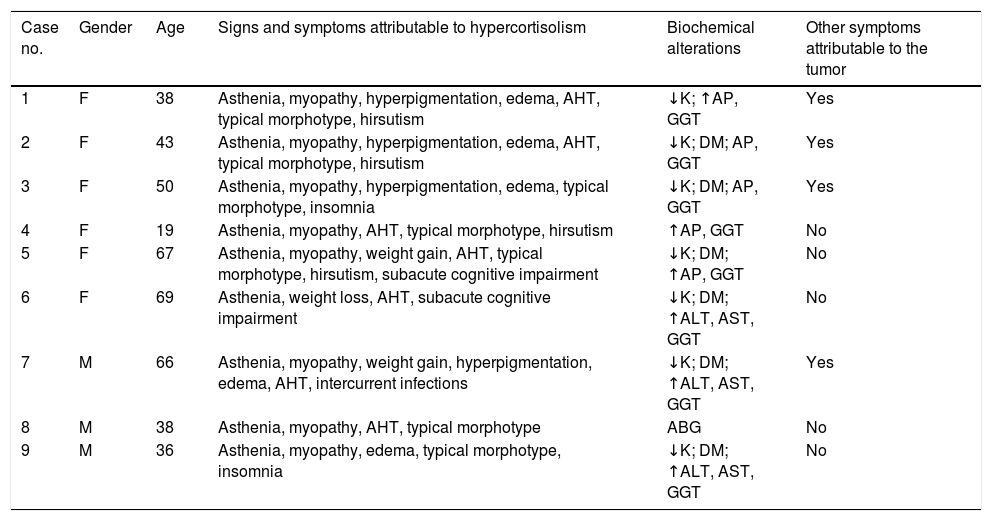
ResultsNine cases of ECS were identified in the period 2000–2015. Of these, 6 were women, and the mean patient age at diagnosis was 47 years (Table 1).
Clinical presentation.
| Case no. | Gender | Age | Signs and symptoms attributable to hypercortisolism | Biochemical alterations | Other symptoms attributable to the tumor |
|---|---|---|---|---|---|
| 1 | F | 38 | Asthenia, myopathy, hyperpigmentation, edema, AHT, typical morphotype, hirsutism | ↓K; ↑AP, GGT | Yes |
| 2 | F | 43 | Asthenia, myopathy, hyperpigmentation, edema, AHT, typical morphotype, hirsutism | ↓K; DM; AP, GGT | Yes |
| 3 | F | 50 | Asthenia, myopathy, hyperpigmentation, edema, typical morphotype, insomnia | ↓K; DM; AP, GGT | Yes |
| 4 | F | 19 | Asthenia, myopathy, AHT, typical morphotype, hirsutism | ↑AP, GGT | No |
| 5 | F | 67 | Asthenia, myopathy, weight gain, AHT, typical morphotype, hirsutism, subacute cognitive impairment | ↓K; DM; ↑AP, GGT | No |
| 6 | F | 69 | Asthenia, weight loss, AHT, subacute cognitive impairment | ↓K; DM; ↑ALT, AST, GGT | No |
| 7 | M | 66 | Asthenia, myopathy, weight gain, hyperpigmentation, edema, AHT, intercurrent infections | ↓K; DM; ↑ALT, AST, GGT | Yes |
| 8 | M | 38 | Asthenia, myopathy, AHT, typical morphotype | ABG | No |
| 9 | M | 36 | Asthenia, myopathy, edema, typical morphotype, insomnia | ↓K; DM; ↑ALT, AST, GGT | No |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; DM: diabetes mellitus; AP: alkaline phosphatase; ABG: altered basal blood glucose; GGT: gamma-glutamyl transferase; AHT: arterial hypertension; K: serum potassium; F: female; M: male.
The most common symptoms at diagnosis were asthenia and muscle weakness as the expression of proximal myopathy (Table 1). Seven patients moreover presented arterial hypertension and weight gain; four had hyperpigmentation, edema, hirsutism and neurocognitive disorders or insomnia; and one case presented treatment-resistant opportunistic infections. All but two patients (cases 6 and 7) presented a characteristic morphotype with centripetal fat distribution, and two of them had skin striae and/or ecchymosis (cases 8 and 9). All patients developed manifest CS, and all except one (case 8) did so in under three months.
Mean basal blood glucose was 185mg/dl (range 84–344), and was in the diabetic range in 6 patients, two of which (cases 5 and 7) had previously known diabetes. Seven patients presented hypopotassemia (mean value: 2.68mmol/l [range 2–3.1]), and 8 showed transaminase elevation (dissociated cholestasis pattern in 5 subjects and mixed in 3).
At the time of presentation, no patient had been diagnosed with neoplastic disease, though four of the 9 subjects had symptoms consistent with such disease and different from those inherent to CS (Zollinger-Ellison syndrome in three cases and dyspnea in another).
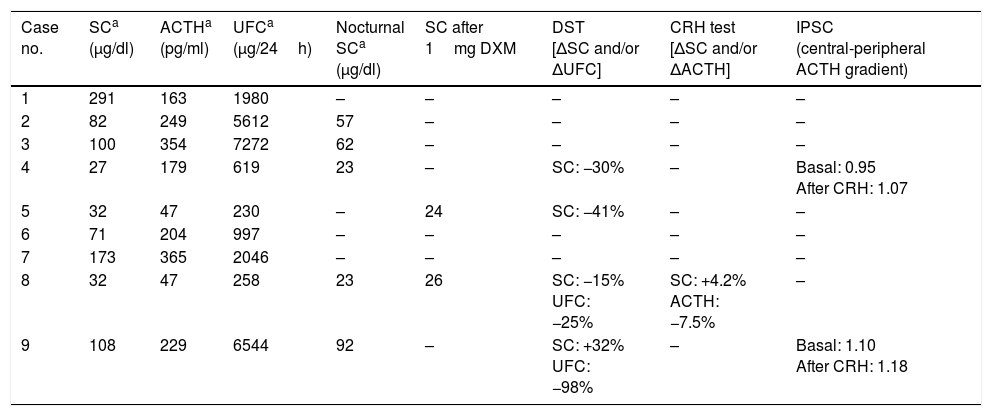
Biochemical diagnosisThe mean baseline SC value was 102μg/dl, with urinary free cortisol 2840μg/24h (Table 2). In all cases, 24-h urinary free cortisol was more than three-fold the upper limit of normal, with a mean of 20-fold the upper limit (range 4.5- to 100-fold). Mean nocturnal SC (n=5) was 51μg/dl, while SC after 1mg DXM (n=2) was 25μg/dl. Mean ACTH concentration was 204pg/ml, with values within the normal range in only two of the 9 patients.
Hormonal parameters.
| Case no. | SCa (μg/dl) | ACTHa (pg/ml) | UFCa (μg/24h) | Nocturnal SCa (μg/dl) | SC after 1mg DXM | DST [ΔSC and/or ΔUFC] | CRH test [ΔSC and/or ΔACTH] | IPSC (central-peripheral ACTH gradient) |
|---|---|---|---|---|---|---|---|---|
| 1 | 291 | 163 | 1980 | – | – | – | – | – |
| 2 | 82 | 249 | 5612 | 57 | – | – | – | – |
| 3 | 100 | 354 | 7272 | 62 | – | – | – | – |
| 4 | 27 | 179 | 619 | 23 | – | SC: −30% | – | Basal: 0.95 After CRH: 1.07 |
| 5 | 32 | 47 | 230 | – | 24 | SC: −41% | – | – |
| 6 | 71 | 204 | 997 | – | – | – | – | – |
| 7 | 173 | 365 | 2046 | – | – | – | – | – |
| 8 | 32 | 47 | 258 | 23 | 26 | SC: −15% UFC: −25% | SC: +4.2% ACTH: −7.5% | – |
| 9 | 108 | 229 | 6544 | 92 | – | SC: +32% UFC: −98% | – | Basal: 1.10 After CRH: 1.18 |
UFC: urinary free cortisol; SC: serum cortisol; IPSC: inferior petrosal sinus catheterization; DST: high-dose dexamethasone suppression test.
Normal values: SC (4.3–22.4μg/dl), ACTH (9–55pg/mL), UFC (11–71μg/24h), nocturnal SC (<7.5μg/dl), SC after 1mg DXM (<1.8μg/dl).
In 5 patients (cases 1, 2, 3, 6 and 7) the ectopic origin was established by markedly elevated urinary free cortisol and ACTH levels, together with radiological evidence of tumor disease potentially capable of explaining the condition. In the remaining four cases, DST was performed as a first approach for establishing the differential diagnosis between CD and ECS. In three of them (cases 4, 5 and 8), all with moderately high urinary free cortisol levels, no SC and/or urinary free cortisol suppression was observed. The ectopic origin of SC was demonstrated by the detection of neuroendocrine tumor metastasis in one patient (case 5), a negative CRH test and the demonstration of a neuroendocrine tumor in another patient (case 8), and by inferior petrosal sinus catheterization with the absence of gradient in the third patient (case 4). The fourth patient subjected to DST (case 9), whose urinary free cortisol levels were markedly elevated, showed urinary free cortisol suppression >90%. The hypophyseal MRI scan revealed a doubtful lesion, though inferior petrosal sinus catheterization confirmed the extrapituitary origin of ACTH secretion.
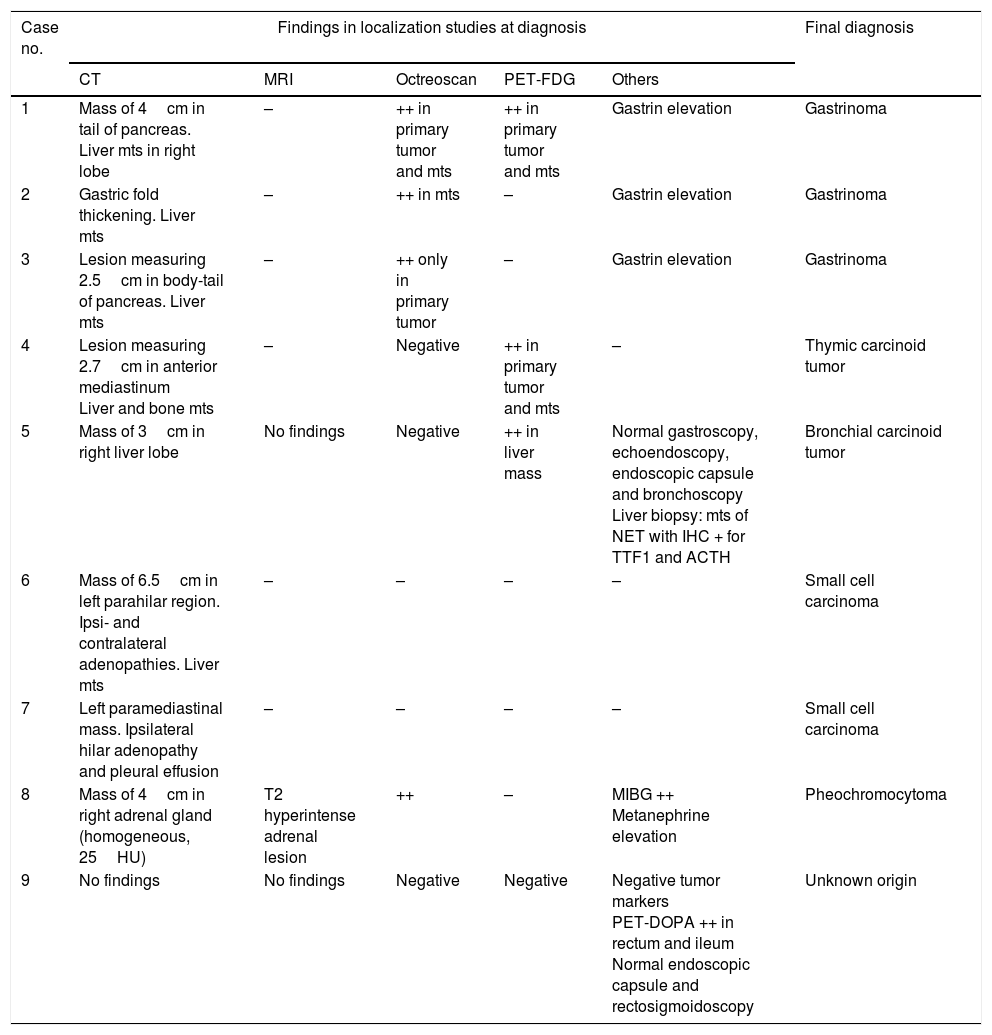
Location of the ACTH sourceImaging studies (CT or MRI) were carried out in all cases: in two of them prior to the diagnosis of ECS (cases 6 and 8), and in the others with the purpose of locating the ectopic ACTH secretion source.
In those patients where the studies were made after the diagnosis of ECS had been confirmed, a lesion potentially causing the syndrome could be identified in four instances (cases 1, 3, 4 and 7), with metastatic spread in all of them (Table 3). In one patient (case 2) multiple liver metastases and gastric fold thickening were identified, which together with the clinical and biochemical data (diarrhea and gastrin>22,000pg/ml [NR<100]) confirmed the diagnosis of gastrinoma. Another patient (case 5) presented a single liver lesion in which the biopsy revealed neuroendocrine tumor metastasis with ACTH-positive immunohistochemical findings. In one patient (case 9), the imaging techniques failed to identify any lesions.
Results of the initial localization studies.
| Case no. | Findings in localization studies at diagnosis | Final diagnosis | ||||
|---|---|---|---|---|---|---|
| CT | MRI | Octreoscan | PET-FDG | Others | ||
| 1 | Mass of 4cm in tail of pancreas. Liver mts in right lobe | – | ++ in primary tumor and mts | ++ in primary tumor and mts | Gastrin elevation | Gastrinoma |
| 2 | Gastric fold thickening. Liver mts | – | ++ in mts | – | Gastrin elevation | Gastrinoma |
| 3 | Lesion measuring 2.5cm in body-tail of pancreas. Liver mts | – | ++ only in primary tumor | – | Gastrin elevation | Gastrinoma |
| 4 | Lesion measuring 2.7cm in anterior mediastinum Liver and bone mts | – | Negative | ++ in primary tumor and mts | – | Thymic carcinoid tumor |
| 5 | Mass of 3cm in right liver lobe | No findings | Negative | ++ in liver mass | Normal gastroscopy, echoendoscopy, endoscopic capsule and bronchoscopy Liver biopsy: mts of NET with IHC + for TTF1 and ACTH | Bronchial carcinoid tumor |
| 6 | Mass of 6.5cm in left parahilar region. Ipsi- and contralateral adenopathies. Liver mts | – | – | – | – | Small cell carcinoma |
| 7 | Left paramediastinal mass. Ipsilateral hilar adenopathy and pleural effusion | – | – | – | – | Small cell carcinoma |
| 8 | Mass of 4cm in right adrenal gland (homogeneous, 25HU) | T2 hyperintense adrenal lesion | ++ | – | MIBG ++ Metanephrine elevation | Pheochromocytoma |
| 9 | No findings | No findings | Negative | Negative | Negative tumor markers PET-DOPA ++ in rectum and ileum Normal endoscopic capsule and rectosigmoidoscopy | Unknown origin |
IHC: immunohistochemistry; MIBG: metaiodobenzylguanidine; Mts: metastases; MRI: magnetic resonance imaging; CT: computed tomography; NET: neuroendocrine tumor; TTF1: thyroid transcription factor 1; HU: Hounsfield units.
Imaging study was carried out in two patients before the diagnosis of ECS was confirmed (cases 6 and 8). In case 6, the study (indicated due to transaminase elevation) showed a lung mass with liver metastases. In case 8, abdominal CT performed during the study of hypercortisolism in another center identified an indeterminate adrenal mass that had not been subjected to further evaluation. After the diagnosis of ECS in our center, the study of this mass revealed pheochromocytoma.
Octreoscan was performed in the three patients lacking evidence of a primary tumor in the anatomical imaging studies (cases 2, 5 and 9), but no additional information was obtained in any of the cases. In turn, FDG-PET was performed in two patients: in one (case 5) the technique identified the already known liver metastatic disease but not the primary tumor, while in the other (case 9) it revealed no lesions. In the latter case, PET-18F-DOPA showed pathological uptake in the rectum and ileum. However, rectosigmoidoscopy and an endoscopic capsule study revealed no lesions.
In addition, octreoscan was performed in four of the 6 cases with locations previously established by CT/MRI. The results proved positive in three patients (cases 1, 3 and 8) and negative in one (case 4).
Overall, the initial ectopic secretion localization tests led to the identification of the primary tumor in 6 cases, while in two cases it was possible to identify metastases but not the primary tumor (cases 2 and 5), and in one patient no lesions could be identified (case 9). Repeat imaging studies during follow-up identified the primary tumor (a 0.9cm lung nodule) 18 months after the initial diagnosis in case 5. By contrast, in case 9 repeat imaging and tumor marker testing failed to identify the primary tumor, which currently remains unknown (Table 3).
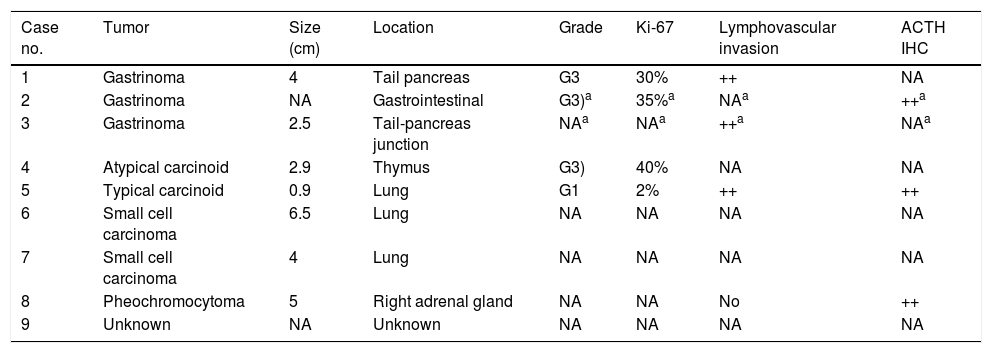
Characteristics of the primary tumorThe primary tumor was identified in 7 cases: two pancreatic gastrinomas, two small cell lung carcinomas, a thymic carcinoid lesion, a bronchial carcinoid tumor, and a pheochromocytoma. In case 2, a diagnosis of gastrinoma was established, without the exact location being identified. The most common sites were gastrointestinal (3 patients) and pulmonary (3 patients). The primary tumor size ranged from 0.9 to 6.5cm, and in 7 cases disseminated disease was found at diagnosis. Immunohistochemical testing for ACTH was performed in three patients and proved positive in all of them (Table 4).
Characteristics of the primary tumor.
| Case no. | Tumor | Size (cm) | Location | Grade | Ki-67 | Lymphovascular invasion | ACTH IHC |
|---|---|---|---|---|---|---|---|
| 1 | Gastrinoma | 4 | Tail pancreas | G3 | 30% | ++ | NA |
| 2 | Gastrinoma | NA | Gastrointestinal | G3)a | 35%a | NAa | ++a |
| 3 | Gastrinoma | 2.5 | Tail-pancreas junction | NAa | NAa | ++a | NAa |
| 4 | Atypical carcinoid | 2.9 | Thymus | G3) | 40% | NA | NA |
| 5 | Typical carcinoid | 0.9 | Lung | G1 | 2% | ++ | ++ |
| 6 | Small cell carcinoma | 6.5 | Lung | NA | NA | NA | NA |
| 7 | Small cell carcinoma | 4 | Lung | NA | NA | NA | NA |
| 8 | Pheochromocytoma | 5 | Right adrenal gland | NA | NA | No | ++ |
| 9 | Unknown | NA | Unknown | NA | NA | NA | NA |
IHC: immunohistochemistry; NA: not available.
The definitive identification of the ACTH source was established in four patients by the demonstration of positive ACTH immunohistochemical staining (cases 2 and 5), the resolution of hypercortisolism after successful tumor treatment (case 1), or both (case 8).
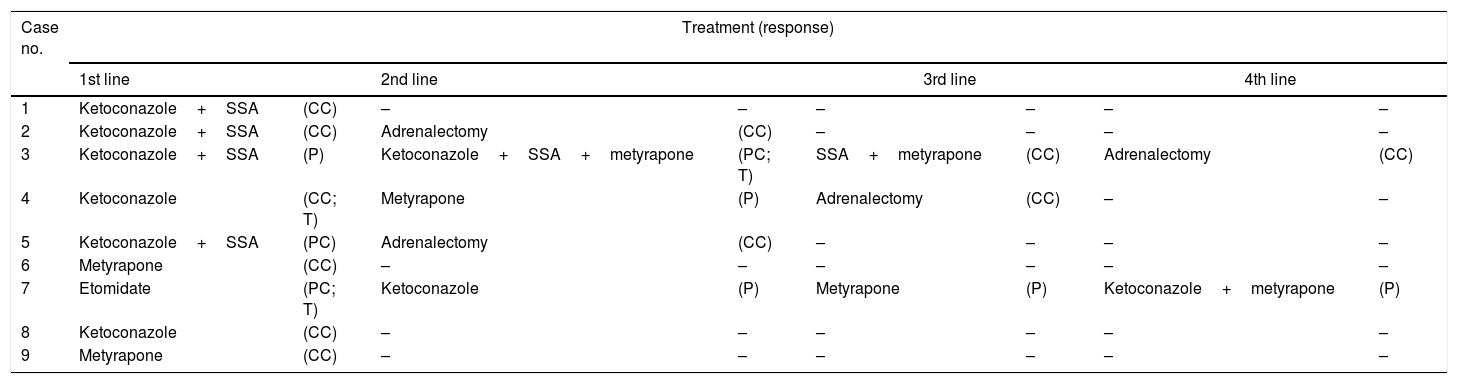
TreatmentTreatment of hypercortisolismAll patients received medical treatment for hypercortisolism before undergoing antitumor treatment (Table 5).
- (a)
Ketoconazole was used as the first measure to control hypercortisolism in 6 cases: in two of them as monotherapy, and combined with somatostatin analogs (lanreotide or octreotide) in the remaining four cases. The two patients subjected to monotherapy (cases 4 and 8) had moderately elevated urinary free cortisol levels, and both achieved normalization with treatment. Of the 4 cases treated with dual therapy (ketoconazole+somatostatin analogs) UFC normalized in 2 cases (1 and 2) and remained elevated in 2 others (3 and 5), one of which had only slightly elevated UFC levels (5).
- (b)
In two patients (cases 1 and 9), metyrapone (Metopirone®) was used as the initial treatment for hypercortisolism due to pre-existing transaminase elevation that precluded the use of ketoconazole. Sustained hypercortisolism control was achieved in both patients despite tumor persistence (case 9) or relapse (case 1).
- (c)
Finally, etomidate was used to initially treat hypercortisolism in one patient (case 7). This patient was admitted to the Intensive Care Unit with orotracheal intubation due to the complications of hypercortisolism. Intravenous administration was therefore required, capable of securing rapid control of the life-threatening hypercortisolism. Etomidate was used in a block-and-replace regimen, a significant decrease in SC levels (from 471 to 149μg/dl) being obtained, but the treatment was discontinued on the sixth day due to a marked increase in plasma osmolarity attributable to the propyleneglycol used as an excipient.
Treatment of hypercortisolism.
| Case no. | Treatment (response) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1st line | 2nd line | 3rd line | 4th line | |||||
| 1 | Ketoconazole+SSA | (CC) | – | – | – | – | – | – |
| 2 | Ketoconazole+SSA | (CC) | Adrenalectomy | (CC) | – | – | – | – |
| 3 | Ketoconazole+SSA | (P) | Ketoconazole+SSA+metyrapone | (PC; T) | SSA+metyrapone | (CC) | Adrenalectomy | (CC) |
| 4 | Ketoconazole | (CC; T) | Metyrapone | (P) | Adrenalectomy | (CC) | – | – |
| 5 | Ketoconazole+SSA | (PC) | Adrenalectomy | (CC) | – | – | – | – |
| 6 | Metyrapone | (CC) | – | – | – | – | – | – |
| 7 | Etomidate | (PC; T) | Ketoconazole | (P) | Metyrapone | (P) | Ketoconazole+metyrapone | (P) |
| 8 | Ketoconazole | (CC) | – | – | – | – | – | – |
| 9 | Metyrapone | (CC) | – | – | – | – | – | – |
ASS: somatostatin analogs; CC: complete control; PC: partial control; P: persistence; T: toxicity.
A second line of treatment was required in 5 patients, because of the persistence or recurrence of hypercortisolism, or due to toxicity. In two of these patients (cases 2 and 5) radical (bilateral) adrenalectomy (RA) was the chosen treatment, while in the remaining three subjects an alternative drug or a combination of several drugs was used. Finally, two of these three patients (cases 3 and 4) underwent RA due to difficulties in controlling hypercortisolism and in order to avoid prolonged exposure to steroidogenic inhibitors. The third patient, treated with etomidate as first line treatment, died within a few days without achieving hypercortisolism control at any time.
Antitumor treatmentSince the disease in most cases was widespread at the time of diagnosis, surgery with curative intent could only be performed in one patient (case 8), with CS remission. Of the 7 cases with metastatic spread (Table 6):
- (a)
Three underwent surgery: in one patient (case 4) resection of the primary tumor was performed in order to facilitate hormone control and to allow for histological confirmation; in another patient (case 5) complete resection of a solitary liver metastasis was performed in a first step, while removal of the primary tumor was performed in a second step, since 18 months elapsed between the identification of the one and the other; and in the third patient (case 1) both the primary tumor and metastasis were resected simultaneously (right hepatectomy). After surgery, chemotherapy (CT) was administered in one patient (case 4) (temozolomide+capecitabine) for tumor control, achieving stable disease for 6 months; in the other two patients (cases 1 and 5), somatostatin analogs were used, achieving stable disease for 45 and 75 months, respectively.
- (b)
Chemotherapy based on the carboplatin-etoposide scheme was used as primary treatment for the disease in three cases. In one patient (case 3) treatment was discontinued after three cycles due to tumor progression, and in two patients (cases 2 and 6) stable disease was achieved for 12 and 5 months after they had received a total of 6 and 4 cycles, respectively.
- (c)
In one patient (case 7) the disease proved highly aggressive, with rapid and profound clinical deterioration that made antitumor treatment impractical, and the patient died 13 days after the diagnosis.
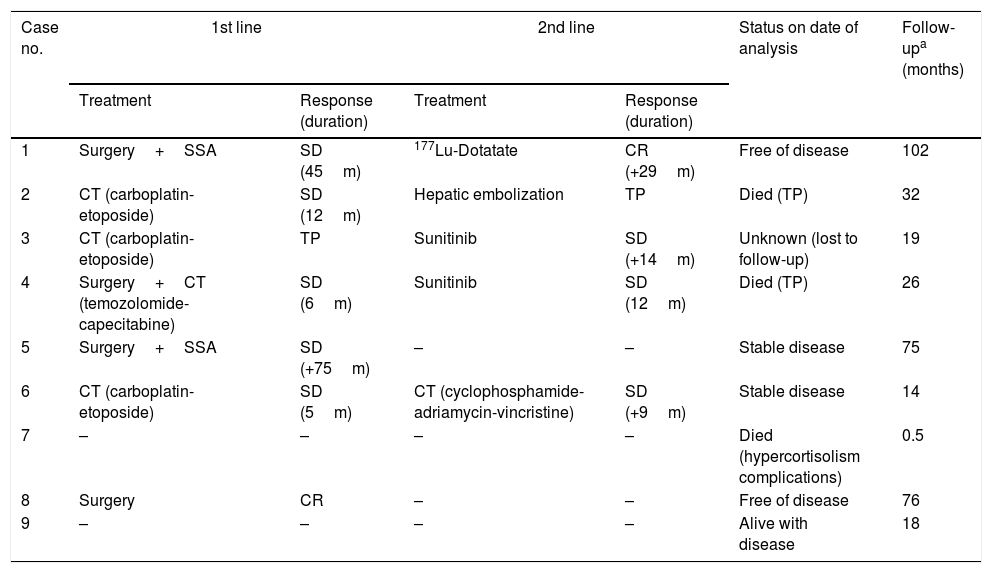
Antitumor therapy and patient status on date of analysis.
| Case no. | 1st line | 2nd line | Status on date of analysis | Follow-upa (months) | ||
|---|---|---|---|---|---|---|
| Treatment | Response (duration) | Treatment | Response (duration) | |||
| 1 | Surgery+SSA | SD (45m) | 177Lu-Dotatate | CR (+29m) | Free of disease | 102 |
| 2 | CT (carboplatin-etoposide) | SD (12m) | Hepatic embolization | TP | Died (TP) | 32 |
| 3 | CT (carboplatin-etoposide) | TP | Sunitinib | SD (+14m) | Unknown (lost to follow-up) | 19 |
| 4 | Surgery+CT (temozolomide-capecitabine) | SD (6m) | Sunitinib | SD (12m) | Died (TP) | 26 |
| 5 | Surgery+SSA | SD (+75m) | – | – | Stable disease | 75 |
| 6 | CT (carboplatin-etoposide) | SD (5m) | CT (cyclophosphamide-adriamycin-vincristine) | SD (+9m) | Stable disease | 14 |
| 7 | – | – | – | – | Died (hypercortisolism complications) | 0.5 |
| 8 | Surgery | CR | – | – | Free of disease | 76 |
| 9 | – | – | – | – | Alive with disease | 18 |
SSA: somatostatin analogs; SD: stable disease; TP: tumor progression; CT: chemotherapy; CR: complete remission; ECS: ectopic Cushing's syndrome.
Tumor progression was observed in four of the 5 patients who achieved stable disease with primary therapy, with a mean time to progression of 17 months. After progression, one of these patients (case 1) received treatment with 177Lu-Dotatate, achieving a complete remission of the disease that was seen to persist on the occasion of the last control (29 months after the end of treatment). In another patient (case 2), embolization was performed of the metastatic lesions in both liver lobes, though disease progression nevertheless occurred and the patient died 32 months after diagnosis due to liver failure. The third patient (case 4) was treated with sunitinib, achieving stable disease for 12 months. Lastly, the fourth patient (case 6) received a second line of chemotherapy with cyclophosphamide+adriamycin+vincristine, maintaining stable disease on the date of the last control (9 months after the end of treatment).
The patient who experienced tumor progression with primary therapy (case 3) received sunitinib as second line treatment, resulting in stable disease during 14 months, after which he was lost to follow-up.
Patient status at end of follow-upAfter a mean follow-up of 40 months, two patients had died as a consequence of tumor progression 32 and 26 months after diagnosis, respectively, while another died due to complications of hypercortisolism 13 days after diagnosis. Two patients were in complete remission, three remained stable, and one had been lost to follow-up (Table 6).
DiscussionEctopic Cushing syndrome is an uncommon condition, as evidenced by this series comprising 9 cases compiled over 15 years in a center with a reference population of over 450,000 inhabitants, and with the potential for recruiting cases from other areas due to the recognized experience of the center in the diagnosis and treatment of CS. Nevertheless, this is the largest series published in Spain to date.
The age and gender distribution was similar to that found in previous series. Clinically, we identified the two extremes of the clinical spectrum of ECS: slowly progressing disease, indistinguishable from CS of pituitary origin (case 8); and disease with an explosive course manifesting with constitutional symptoms and an absence of the classical morphotype (case 7). However, most patients presented data suggesting ectopic ACTH secretion, such as edema or hypopotassemia. These signs reflect the existence of markedly elevated cortisol, capable of accessing the renal tubular mineralocorticoid receptors by overwhelming the inactivating capacity of the enzyme β-hydroxysteroid dehydrogenase type 2. While these signs are not exclusive to ECS, they are very rare in CS of other origin.5,10–14
Six of the 9 patients had markedly higher SC, UFC and ACTH levels than those commonly found in CD, and one additional patient had clearly elevated ACTH. Only two patients presented SC, UFC and ACTH levels within ranges similar to those seen in CD (though the considerable overlap in the values of these measurements in patients with ECS and CD must be taken into account), and neither exhibited clinical data suggestive of ECS. In fact, one of these two patients (described in detail in another publication)15 had been misdiagnosed with CD and underwent pituitary surgery before reporting to our center. The occult tumor ACTH levels exceeded those seen in four patients with evident tumors, thus confirming the lack of correlation between ACTH levels and tumor size reported in other studies.14
The overlapping of clinical and hormonal data between CD and ECS, described in up to 30% of all cases, requires additional tests in order to differentiate the disorders.1,10,12,16–18 The high-dose dexamethasone suppression test (DST) is highly specific for identifying CD when UFC suppression exceeds 90%, though the absence of suppression is scantly specific of ECS.19 In turn, inferior petrosal sinus catheterization (IPSC) has a very high sensitivity and specificity, and is the test of choice used in many centers, though it is an invasive procedure involving risks. Therefore, IPSC should be reserved for patients with suspect clinical and/or biochemical data or with noninvasive test findings consistent with an ectopic origin in which there is no radiological evidence of a tumor lesion.20–22 However, often because of the severity of the clinical condition, requiring early treatment measures, and the risks associated with some of these tests, such studies are not made in patients with ECS, as was shown in our series.
With the exception of medullary thyroid carcinoma, all the tumor lines associated with ectopic ACTH secretion are represented in our series despite its small size. Although small cell lung cancer and bronchial carcinoid tumors are usually the predominant presentations,5,6,10–12 gastrinoma was the most prevalent lesion in our study, affecting three of the 9 patients, as compared to approximately 15% in most series. This difference is probably attributable to chance, since we have been unable to find any other explanation.
In 8 patients (89%) the tumor or its metastases could be identified by conventional imaging techniques performed during or shortly after the diagnosis of ECS. This localization rate is higher than reported in other series, with figures of approximately 50%,23 though it should be noted that in our series almost one-half of the patients presented symptoms other than those derived from CS (diarrhea and dyspnea), and most of the cases presented advanced tumor stages. Scintigraphy with somatostatin analogs may occasionally be helpful for identifying tumors not identified by CT/MRI,5,23–26 though this did not occur in the only patient in our series with no evidence of tumor disease in the anatomical imaging explorations. As is usual, PET-FDG likewise did not contribute information in this regard, because occult tumors usually have a low metabolic rate.26 Positron emission tomography with 18F-DOPA, a radiotracer more specific of neuroendocrine tumors, revealed uptake in the ileum and rectum, but the endoscopic studies showed no abnormalities in these locations. It is therefore likely that this constituted a false-positive PET-18F-DOPA result, though the specificity of this technique as recorded to date has been 100%.23 Tumor markers (gastrin, calcitonin, metanephrines), which can help locate the source of ACTH,1–3,5 were likewise not useful on this occasion. The ACTH source thus remains unknown in this patient, a situation that occurs in approximately 15% of all cases of ECS.5,11,24,27 Although repeatedly failed attempts to find the tumor indicate a favorable prognosis, this does not guarantee the absence of malignancy.4,5,20,21 Long-term follow-up is therefore mandatory in these cases.
The time relationship between the diagnosis of ECS and the diagnosis of the tumor varies, though it is common for ECS to manifest in the context of tumor progression or relapse.1–5,12,16 In agreement with this, 7 of the 9 cases reported in our series had metastatic spread at the time of diagnosis. However, no patient had been diagnosed with neoplastic disease when CS was suspected. This is probably due to the low specificity of the symptoms, which prevents their tumor origin from being recognized, as occurred in the three gastrinomas. In these patients, the onset of Zollinger-Ellison syndrome occurred at least one year before the start of CS.
The only curative treatment for ECS is complete resection or elimination of the neoplasm. However, given the frequent metastatic spread found in our series, healing was only achieved in two of the 9 patients, this being similar to the 10–15% rate reported in the literature.28
In poorly differentiated tumors, the treatment alternative of choice is platinum-based chemotherapy, while in well-differentiated tumors, somatostatin analogs, chemotherapy, mTOR (everolimus) or tyrosine kinase inhibitors (sunitinib), and radionuclide therapy, have been shown to be useful, with variable response rates.29 The existing evidence does not allow for the definition of the initial treatment of choice or of the most appropriate treatment sequence after tumor progression. Recently, radionuclide treatment (177Lu-Dotatate) in subjects with non-resectable middle bowel neuroendocrine tumors progressing after somatostatin analog therapy has yielded a radiological response rate and a duration of response superior to that of any other therapeutic modality.30 In accordance with this, the only patient in our series treated with 177Lu-Dotatate achieved complete and sustained clinical and radiological response over time.
The control of hypercortisolism through medical treatment is mandatory before the patient is subjected to surgery or chemotherapy, because it significantly decreases the morbidity, mortality and toxicity related to such therapies and increases the response rate.31 Also, such control is a key element in the management of ECS when tumor remission is not possible. Ketoconazole and metyrapone (Metopirone®) (sometimes associated with somatostatin analogs) were effective in most of our cases.
Because of its almost immediate effect and the possibility of intravenous administration, etomidate should be reserved for critical situations derived from life-threatening hypercortisolism.32 The hyperosmolarity associated with etomidate therapy is a surrogate marker of propyleneglycol levels,33 used as a vehicle in some drug formulations. High concentrations of this compound have been associated with toxicity and the need for treatment discontinuation; the use of propyleneglycol-free formulations (if available) is therefore to be preferred. Radical (bilateral) adrenalectomy (RA) is the only possible treatment in cases not controlled by medical treatment or when toxicity develops, provided life expectancy is sufficiently prolonged as a result. Radical adrenalectomy is also indicated when the avoidance of prolonged exposure to steroidogenic inhibitors is being sought, and in the case of occult tumors when the chances of tumor localization are small.34
The prognosis of ECS is determined by the type of tumor involved, the severity of hypercortisolism,12,16,24 and the speed and effectiveness with which it is controlled.3,5,12 In addition, the prognosis is more favorable in patients with occult tumors than in patients with an evident ACTH source. In our series, after a mean follow-up of 40 months, only two patients were in complete remission. This is in concordance with the data found in the literature.10 Three patients died in a relatively short time interval after diagnosis (mean 19.5 months), all of them with tumor lines recognized as having a poor prognosis, and with severe hypercortisolism that proved difficult to control.
In conclusion, our study found ECS to be an infrequent disorder, associated with a broad spectrum of neuroendocrine tumors of different aggressivity and nature. The ectopic origin of CS can usually be suspected from clinical and biochemical data, and in most cases can be confirmed without the need for invasive tests. The disorder is indicative of poor prognosis, since it is usually diagnosed when the tumor is in advanced stages.
Multiple treatment modalities were required for controlling both the hypercortisolism and the tumor. Antitumor treatment resulted in the cure or the stabilization of the neoplastic disease in over half of the patients, and hypercortisolism control proved possible after medical and/or surgical treatment in all but one patient. Given the complexity of the condition, we recommend the intervention of staff experienced in oncology and endocrinology in order to ensure adequate management and to improve the prognosis.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Araujo Castro M, Palacios García N, Aller Pardo J, Izquierdo Alvarez C, Armengod Grao L, Estrada García J. Síndrome de Cushing ectópico: descripción de 9 casos. Endocrinol Diabetes Nutr. 2018;65:255–264.
