





Epistasis is a type of genetic interaction that could explain much of the phenotypic variability of complex diseases. In this work, the effect of epistasis of metabolic genes and cardiovascular risk on the susceptibility to the development of ischaemic heart disease in Yucatan was determined.
MethodsCase–control study in 79 Yucatecan patients with ischaemic heart disease and 101 healthy controls matched by age and origin with cases. The polymorphisms -108CT, Q192R, L55 M (paraoxonase 1; PON1), C677T, A1298C (5,10-methylenetetrahydrofolate reductase; MTHFR), and the presence/absence of the glutathione S-transferase T1 (GSTT1) gene were genotyped. Epistasis analysis was performed using the multifactorial dimensional reduction method. The best risk prediction model was selected based on precision (%), statistical significance (p<0.05) and cross-validation consistency.
ResultsWe found an independent association of the null genotype GSTT1*0/0 (OR=3.39; CI: 1.29–8.87; p=0.017) and the null allele (OR=1.86; CI: 1.19–2.91; p=0.007) with ischaemic heart disease. The GSTT1*0 deletion and the 677TT genotype (MTHFR) were identified as being at a high cardiovascular risk, whereas the GSTT1*1 wild type genotype and the CC677 variant were at low risk. The gene–environment interaction identified the GSTT1 gene, C677T polymorphism (MTHFR) and hypertension as the factors that best explain ischaemic heart disease in the study population.
ConclusionsThe interaction of the GSTT1 and MTHFR and hypertension may constitute a predictive model of risk for early onset ischaemic heart disease in the population of Yucatan.
La epistasia es un tipo de interacción genética que podría explicar gran parte de la variabilidad fenotípica que muestran las enfermedades complejas. En este trabajo se determinó el efecto de la epistasia de genes metabólicos y los factores de riesgo cardiovascular en la susceptibilidad al desarrollo de cardiopatía isquémica en Yucatán.
MétodosEstudio de casos y controles en 79 pacientes yucatecos con cardiopatía isquémica y 101 controles sanos pareados por edad y origen con los casos. Se genotipificaron los polimorfismos -108CT, Q192R, L55M (paraoxonasa 1, PON1), C677T, A1298C (5,10 metilentetrahidrofolato reductasa, MTHFR) y la presencia/ausencia del gen glutatión S-transferasa T1 (GSTT1). El análisis de epistasia se realizó con el método de reducción dimensional multifactorial (MDR). El mejor modelo de predicción de riesgo se seleccionó con base en la precisión (%), la significación estadística (p<0,05) y la consistencia de la validación cruzada.
ResultadosSe encontró asociación independiente del genotipo nulo GSTT1*0/0 (OR=3,39; IC: 1,29-8,87; p=0,017) y el alelo nulo (OR=1,86; IC: 1,19-2,91; p=0,007) con la cardiopatía isquémica. La deleción GSTT1*0 y el genotipo 677TT (MTHFR) se identificaron de alto riesgo cardiovascular, mientras que el genotipo silvestre GSTT1*1 y la variante CC677 se clasificaron de bajo riesgo. La interacción gen-ambiente identificó al gen GSTT1, al polimorfismo C677T (MTHFR) y a la hipertensión arterial como los factores que mejor explican la cardiopatía isquémica en la población estudiada.
ConclusionesLa interacción de los genes GSTT1 y MTHFR conjuntamente con la hipertensión arterial puede constituir un modelo de predicción de riesgo para el inicio temprano de cardiopatía isquémica en la población de Yucatán.
Ischaemic heart disease, or coronary artery disease (CAD), is a complex disease with inheritance patterns formed by combinations of genes belonging to multiple loci in interaction with environmental risk factors. Genetic association studies have traditionally focused on polymorphisms isolated from a single nucleotide (SNP – simple nucleotide polymorphisms). However, it has been difficult to establish an actual association with the disease as it is polygenic in nature.1,2 One of the current strategies for CAD is to identify groups of functional polymorphisms in candidate genes involved in cardiovascular homeostasis which could more precisely define the propensity to a certain atherothrombotic phenotype.3
Epistasis is a type of genetic interaction that could explain much of the phenotypic variability in coronary disease. Multifactor dimensionality reduction (MDR), a method designed for case–control association studies with small sample sizes, is a novel strategy for the identification of epistatic effects which are predictors of a given clinical situation.4,5 MDR is a non-parametric method that reduces the multifactor dimension by grouping the different genotypes into 2 groups: high and low risk. MDR does not assume a specific inheritance model but, once it selects the most relevant polymorphisms, it examines the proportion of cases and controls of each of the genotype combinations between two or more loci to obtain a final classification model for high and low risk. These interactions are represented in a contingency table where for each cell the proportion of cases/controls is estimated. Cells with a case/control ratio equal to or greater than the overall case/control ratio for the population are classified as high risk and those with a smaller ratio than the overall, as low risk. This provides us with a classification model in which high-risk subjects are classified as cases and low-risk subjects as controls. The program generates different interaction models between the loci evaluated, identifying the best in terms of sensitivity and specificity, referred to as balanced accuracy. A cross-validation counts the number of times a certain interaction is chosen as outstanding and thus identifies the best model. A permutation test evaluates whether or not said model is statistically significant.6,7 The best model will be the one with the highest accuracy and significance value (p<0.05).
For this study we selected six polymorphisms of a single nucleotide of the candidate genes glutathione S-transferase T1 (GSTT1), paraoxonase 1 (PON1) and 5,10 methylenetetrahydrofolate reductase (MTHFR) related to antioxidant and folate metabolism, these being processes involved in the pathophysiology of coronary disease of atherosclerotic origin.
The GSTT1 gene codes for enzymes involved in cell detoxification. Wild-type variant GSTT1*1 is functional while null variant GSTT1*0 involves the deletion of the gene.8 The subjects with the homozygous GSTT1*0/*0 deletion lack the enzyme, while the heterozygotes GSTT1*1/*0 have intermediate activity due to the effect of the functional allele.9,10
The PON1 gene codes for a glycoprotein with antioxidant activity associated with HDL molecules (high density lipoproteins) whose concentration increases in response to oxidative stress.11 The variants being examined in this study are located in the promoter region (-108CT [rs705379]) and in the coding region (T260A [rs854560] and A672G [rs662]), the latter known in the literature as L55M and Q192R respectively. These isoforms have different capacities for protecting LDL from lipid oxidation: genotypes 192QQ and 55MM give HDL greater antioxidant activity, with an atheroprotective effect, unlike the atherogenic genotypes 192RR and 55LL.12,13 Meanwhile, the isoform encoded by the -108C allele has been associated with a higher enzyme concentration compared to the -108T14 variant.
Homocysteine (Hcy) is an intermediate product of methionine metabolism, the increase in which is inversely related to plasma levels of folate and considered an atherogenic risk factor, as it promotes endothelial dysfunction.15,16 The most studied polymorphisms of the MTHFR gene are C677T and A1298C. The homozygotes for the T allele (C677T) have lower levels of enzyme activity, associated with low levels of folate and high levels of Hcy,17,18 while the variant A1298C has been associated with low-level enzyme activity in vitro and in vivo.19
The aim of this study was to determine the epistasis effects of six metabolic gene polymorphisms and their interaction with cardiovascular risk factors, and to identify a risk prediction model for CAD in Yucatecan patients.
MethodsType of study and samplesWe designed a case–control study with matching by age and place of birth. We recruited 79 patients, males and females, of Yucatecan descent and with a diagnosis of CAD, from the cardiology departments of the Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado (ISSSTE) [Institute of Social Security and Social Services for Civil Servants] Mérida Regional Hospital and the Dr. Agustín O’Horan General Hospital in Mérida (Yucatán). The diagnostic criteria for CAD (American College of Cardiology Foundation [ACCF]/American Heart Association [AHA])20,21 were central chest pain at rest or on exercise lasting more than 20min, with or without changes on the baseline electrocardiogram at rest, with or without alteration of enzyme markers, diagnosis of which was confirmed by cardiac catheterisation. The controls were 101 apparently healthy donors, unrelated, with no clinical signs of CAD, with stress test negative for myocardial ischaemia and no family history of premature coronary disease (coronary event in first or second degree family members before age 55 in males or 65 in females), from the State Blood Transfusion Centre and the ISSSTE Mérida Regional Hospital.
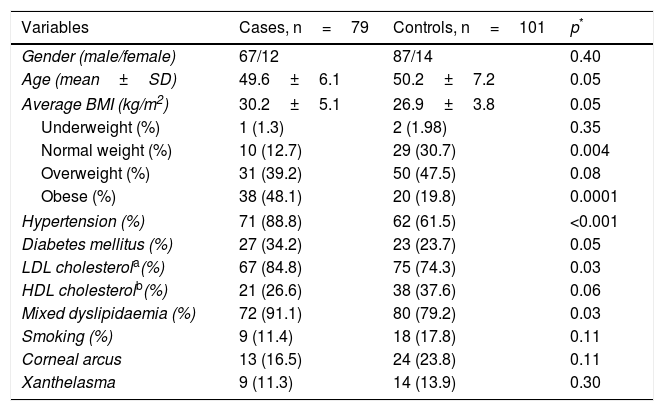
Demographic data and personal history of the cardiovascular risk factors obesity, hypertension, diabetes mellitus (DM), high-density lipoprotein (HDL) cholesterol, low density lipoprotein (LDL) cholesterol and mixed dyslipidaemia were recorded, as well as the presence of corneal arcus and xanthelasma. Body mass index (BMI) was calculated as weight in kilograms divided by height in square metres and categorised as underweight (<18.5), normal weight (18.5 to 24.9), overweight (25.0 to <30.0) and obese (>30.0). Hypertension was defined according to AHA (American Heart Association) recommendations, with systolic/diastolic blood pressure values ≥140/90mmHg.22 Mixed dyslipidaemia was considered when total plasma cholesterol and triglyceride values were above 200mg/dl23 LDL cholesterol was calculated using the Friedewald formula. DM was defined according to the American Diabetes Association as fasting plasma blood glucose ≥126mg/dl, plus classic symptoms of diabetes or blood glucose ≥200mg mg/dl 2h after the oral glucose tolerance test.24 Smoking was defined as whether a person was a smoker or a non-smoker. A person was considered to be a smoker if they said that they had smoked at least one cigarette a day for the last year or more, or that they were living on a daily basis with smokers. They were considered to be non-smokers if they had never smoked or lived with smokers. Among the cases, each category was defined based on the diagnosis of CAD.
All the individuals were defined ethnically as mixed race and stated that they had been born in Yucatan and were descendants of Yucatecan parents and grandparents. The absence of genetic substructure or stratification between the study groups was determined previously, using four ancestral polymorphisms which characterise the ABO*O blood group locus. The study was approved by the Independent Ethics Committees of the participating institutions. Informed consent was obtained from all subjects.
Obtaining of DNA and genotypingGenomic DNA was obtained from 2ml of peripheral blood by a saline precipitation method described by Bunce25 and stored at −20°C until processing.
Polymorphisms C677T and A1298C (MTHFR) were identified by polymerase chain reaction with restriction fragment length polymorphism analysis (PCR-RFLP). For C677T (rs1801133) the oligonucleotides 5′CGAAGCAGGGAGCTTTGAGGCTG3′ (sense) and 5′AGGACGGTGCGGTGAGAGTG3′ (antisense) were used. PCR conditions were: initial denaturation at 94°C for 5min, followed by 35 cycles, 30s at 94°C, 45s at 62°C and 30s at 72°C, with final extension at 72°C for 5min. The amplified product of 233bp was treated with the restriction enzyme TaqI (Thermo Scientific) for 1h at 37°C. The restriction products were separated by 12% polyacrylamide gel electrophoresis with silver nitrate. The enzyme produces a single cut-off site and generates a fragment of 171bp corresponding to the T allele. For A1298C (rs1801131), we used the oligonucleotides (sense) 5′-CTTTGGGGAGCTGAAGGACTACT-3′ and (antisense) 5′-CACTTTGTGACCATTCCGGTTTG-3′. The amplification conditions were: initial denaturation for 10min at 95°C followed by 35 cycles of 1min at 95°C, 30s of alignment at 62°C, 72°C for 1min and a final extension at 72°C for 7min. The amplified product of 163bp was digested with the restriction enzyme MboII (Thermo Scientific). The products obtained were separated by electrophoresis in 12% polyacrylamide gel with silver nitrate. The MboII enzyme generates fragments of 56bp and 30/31bp for the A allele, and of 84bp and 30/31bp for the C allele.
The genotyping of the GSTT1 gene was performed by multiple PCR according to the protocol described by Buchard et al.26 Two primer pairs were used: one recognises a sequence within the 969bp gene and the second, flanking its ends. If there is a deletion, a fragment of 3106bp will be obtained. PCR conditions were: initial denaturation at 94°C for 3min, followed by 35 cycles, 30s at 94°C, 7min at 68°C and extension at 69°C for 10min. The amplified products were visualised on 1.5% agarose gel with SYBR safe DNA (Invitrogen) by electrophoresis.
Genotyping of PON1 polymorphisms was performed by real-time PCR by allelic discrimination assay with TaqMan probes (Applied Biosystems) -108CT (C_11708905_10), Q192R (C_2548962_20) and L55M (C_2259750_20).
Statistical analysisContinuous variables were expressed as mean±standard deviation and discrete variables as percentages. For each polymorphism, genotype frequencies were estimated by direct counting and allele frequencies from the genotypes, expressed as percentages. The Chi-square test was used to compare the categorical variables and to evaluate the Hardy–Weinberg equilibrium. The odds ratio (OR) and its 95% confidence intervals (CI) were estimated to determine the association of each independent polymorphism with the disease, taking as reference the genotype and the wild-type allele (codominant inheritance model) of each variant, using Stata/SE 12.0. The linkage disequilibrium (LD′) between pairs of SNP and the Pearson correlation coefficient (r2) were evaluated with the SNPstat program (https://www.snpstats.net/snpstats/start.htm). The epistasis analysis was performed with the MDR 3.0.2 program (http://epistasis.org), analysing the six polymorphisms together. MDR was also applied to determine the gene–environment interaction and to identify the weight of each of the independent variables (polymorphisms, hypertension, mixed dyslipidaemia, LDL cholesterol, HDL cholesterol, obesity [BMI >30] and smoking) in relation to the disease. A significance level of p<0.05 was considered for all the analyses.
ResultsThe demographic and clinical characteristics of the patients and the controls are summarised in Table 1. The proportion of males and females and the average age, overall and by gender (male cases 49.9±6.5 vs controls 46.2±7.3, p=0.29; female cases 50.5±6.5 vs controls 46.0±8.4, p=0.29) were similar in both groups. The cardiovascular risk factors hypertension (p<0.001) and mixed dyslipidaemia (p=0.01) were significantly more prevalent in the patient group. When stratifying BMI, we found a higher percentage of controls with normal weight (30.7 vs 12.7%; p=0.004) and of patients with obesity (48.1 vs 19.8%; p=0.0001). Being overweight was the most prevalent factor in the general population, with a slight increase in the control subjects (cases 39.2% and controls 47.5%, p=0.07). The presence of corneal arcus and xanthelasma was similar in both groups (p>0.05).
Anthropometric and clinical characteristics.
| Variables | Cases, n=79 | Controls, n=101 | p* |
|---|---|---|---|
| Gender (male/female) | 67/12 | 87/14 | 0.40 |
| Age (mean±SD) | 49.6±6.1 | 50.2±7.2 | 0.05 |
| Average BMI (kg/m2) | 30.2±5.1 | 26.9±3.8 | 0.05 |
| Underweight (%) | 1 (1.3) | 2 (1.98) | 0.35 |
| Normal weight (%) | 10 (12.7) | 29 (30.7) | 0.004 |
| Overweight (%) | 31 (39.2) | 50 (47.5) | 0.08 |
| Obese (%) | 38 (48.1) | 20 (19.8) | 0.0001 |
| Hypertension (%) | 71 (88.8) | 62 (61.5) | <0.001 |
| Diabetes mellitus (%) | 27 (34.2) | 23 (23.7) | 0.05 |
| LDL cholesterola(%) | 67 (84.8) | 75 (74.3) | 0.03 |
| HDL cholesterolb(%) | 21 (26.6) | 38 (37.6) | 0.06 |
| Mixed dyslipidaemia (%) | 72 (91.1) | 80 (79.2) | 0.03 |
| Smoking (%) | 9 (11.4) | 18 (17.8) | 0.11 |
| Corneal arcus | 13 (16.5) | 24 (23.8) | 0.11 |
| Xanthelasma | 9 (11.3) | 14 (13.9) | 0.30 |
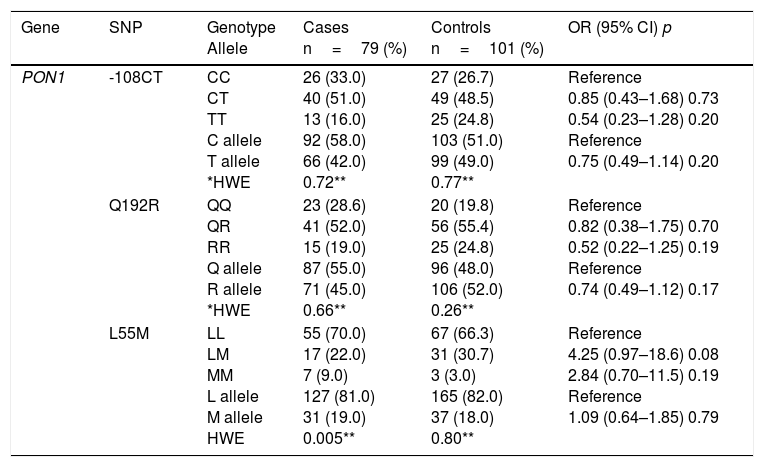
Table 2 shows the comparison of genotype and allele frequencies for the polymorphisms studied in cases and controls, the Hardy–Weinberg equilibrium and the association with CAD according to the co-dominant inheritance model. With the exception of GSTT1, for which a higher prevalence of patients was detected with the homozygous deletion of the gene (GSTT1*0/0), the rest of the polymorphisms did not show significant differences in the genotype and allele frequencies between the two groups (p>0.05). For all the variants, the frequencies observed in the control subjects were found in Hardy–Weinberg equilibrium (p>0.05). The imbalance observed for the L55M polymorphism (p=0.005) in the patient group was not considered relevant, because this bias could be indicative of a causal relationship with the disease.
Genotype and allele frequencies of the studied polymorphisms, Hardy–Weinberg equilibrium and risk estimation (OR) in cases and controls.
| Gene | SNP | Genotype Allele | Cases n=79 (%) | Controls n=101 (%) | OR (95% CI) p |
|---|---|---|---|---|---|
| PON1 | -108CT | CC CT TT C allele T allele *HWE | 26 (33.0) 40 (51.0) 13 (16.0) 92 (58.0) 66 (42.0) 0.72** | 27 (26.7) 49 (48.5) 25 (24.8) 103 (51.0) 99 (49.0) 0.77** | Reference 0.85 (0.43–1.68) 0.73 0.54 (0.23–1.28) 0.20 Reference 0.75 (0.49–1.14) 0.20 |
| Q192R | QQ QR RR Q allele R allele *HWE | 23 (28.6) 41 (52.0) 15 (19.0) 87 (55.0) 71 (45.0) 0.66** | 20 (19.8) 56 (55.4) 25 (24.8) 96 (48.0) 106 (52.0) 0.26** | Reference 0.82 (0.38–1.75) 0.70 0.52 (0.22–1.25) 0.19 Reference 0.74 (0.49–1.12) 0.17 | |
| L55M | LL LM MM L allele M allele HWE | 55 (70.0) 17 (22.0) 7 (9.0) 127 (81.0) 31 (19.0) 0.005** | 67 (66.3) 31 (30.7) 3 (3.0) 165 (82.0) 37 (18.0) 0.80** | Reference 4.25 (0.97–18.6) 0.08 2.84 (0.70–11.5) 0.19 Reference 1.09 (0.64–1.85) 0.79 |
| GSTT1 | 1/1 1/0 0/0 Presence Null HWE | 31 (39.0) 33 (42.0) 15 (19.0) 95 (60.0) 63 (40.0) 0.25a | 56 (55.4) 37 (36.6) 8 (7.9) 149 (74.0) 53 (26.0) 0.59a | Reference 2.10 (0.79–5.59) 0.15 3.39 (1.29–8.87) 0.017 Reference 1.86 (1.19–2.91) 0.007 | |
| MTFFR | C677T | C/C C/T T/T C allele T allele HWE | 18 (23.0) 42 (53.0) 19 (24.0) 78 (49.0) 80 (51.0) 0.57a | 25 (25.0) 41 (41.0) 35 (35.0) 92 (46.0) 110 (54.0) 0.07a | Reference 0.53 (0.26–1.07) 0.08 0.75 (0.33–1.72) 0.53 Reference 0.86 (0.57–1.30) 0.52 |
| A1298C | A/A A/C C/C A allele C allele HWE | 56 (71.0) 22 (28.0) 1 (10.0) 134 (85.0) 24 (15.0) 0.47a | 77 (55.4) 21 (36.6) 3 (7.9) 175 (87.0) 27 (13.0) 0.30a | Reference 0.32 (0.31–3.31) 0.60 0.46 (0.05–4.52) 0.64 Reference 1.16 (0.64–2.10) 0.65 |
HWE: Hardy–Weinberg equilibrium.
The association analysis for each independent polymorphism detected that both the genotype and the null allele of the GSTT1 gene confer greater susceptibility to CAD (3.39 and 1.86 times respectively) compared to subjects who are carriers of the genotype and the wild-type allele. The rest of the polymorphisms showed no risk association.
A high degree of linkage disequilibrium was detected between nucleotides 677 and 1298 of the MTHFR gene (LD′=0.858, r2=0.369, p≤0.0001) and to a lesser extent between Q192R and L55M (LD′=0.394; r2=0.207, p≤0.0001), between -108CT and Q192R (LD′=0.312, r2=0.282, p≤0.0001) and between -108CT and L55M (LD′=0.394, r2=0.207; p≤0.0001) of the PON1 gene. The GSTT1 gene and the C677T variant showed moderate linkage disequilibrium (LD′=0.133, r2=0.087). However, based on the coefficients of correlation and association effects, none of the polymorphisms can be substituted for each other. No risk association with CAD was found based on haplotype estimation (data not shown).
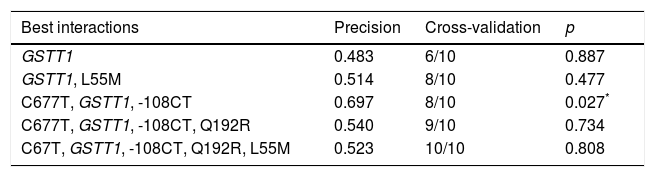
For the multilocus analysis (MDR) the three genotypes were evaluated separately for each polymorphism (co-dominant inheritance model). MDR identified the interaction between the GSTT1 gene and the C677T polymorphisms of the MTHFR gene, and -108CT of PON1 with a precision of 69.7%, a significance of p=0.027 and a cross-validation of 8/10 as the best prediction model. The models with two, four and five interactions showed a decrease in accuracy, although not statistically significant (Table 3). Of the 24 estimated genotype combinations, individuals with deletion of the GSTT1 gene (null genotype) and homozygotes for the T allele (C677T), regardless of the -108CT genotype, were identified in the high cardiovascular-risk group. The model considered high risk as when the ratio of the percentage of cases to controls was higher than 1.25 (Fig. 1).
Summary of the best gene–gene interaction combinations obtained with the MDR analysis.
| Best interactions | Precision | Cross-validation | p |
|---|---|---|---|
| GSTT1 | 0.483 | 6/10 | 0.887 |
| GSTT1, L55M | 0.514 | 8/10 | 0.477 |
| C677T, GSTT1, -108CT | 0.697 | 8/10 | 0.027* |
| C677T, GSTT1, -108CT, Q192R | 0.540 | 9/10 | 0.734 |
| C67T, GSTT1, -108CT, Q192R, L55M | 0.523 | 10/10 | 0.808 |
 high-risk (grey bars) and low-risk (white bars) genotype combinations, corresponding to the three polymorphisms identified in the MDR analysis as the best interaction model (GSTT1/C677T/-108CT). The grey bars represent the cases and the white bars represent the controls.")
Distribution of statistically significant (p<0.005) high-risk (grey bars) and low-risk (white bars) genotype combinations, corresponding to the three polymorphisms identified in the MDR analysis as the best interaction model (GSTT1/C677T/-108CT). The grey bars represent the cases and the white bars represent the controls.
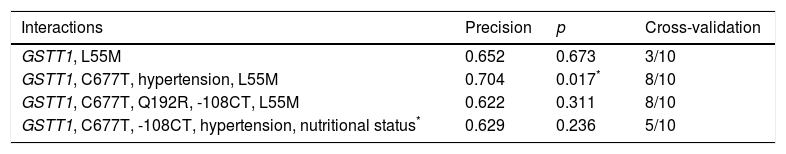
By including MDR (gene–environment interaction) of the risk factors obesity, hypertension, mixed dyslipidaemia, HDL cholesterol, LDL cholesterol, DM, corneal arcus and xanthelasma in the analysis, the best prediction model (p=0.017) maintained the interaction between the GSTT1 gene and the C677T polymorphism (MTHFR), and identified hypertension as the risk factor that contributes most to cardiovascular risk in this population (Table 4).
Best gene–environment interaction models estimated with MDR analysis.
| Interactions | Precision | p | Cross-validation |
|---|---|---|---|
| GSTT1, L55M | 0.652 | 0.673 | 3/10 |
| GSTT1, C677T, hypertension, L55M | 0.704 | 0.017* | 8/10 |
| GSTT1, C677T, Q192R, -108CT, L55M | 0.622 | 0.311 | 8/10 |
| GSTT1, C677T, -108CT, hypertension, nutritional status* | 0.629 | 0.236 | 5/10 |
BMI was considered a categorical variable of nutritional status.
Advanced statistical instruments that consider both the effect of multiple genetic interactions and the environment are required to establish risk prediction models for complex diseases. MDR analysis has already been used in association studies of complex diseases such as cancer,27 type 2 diabetes,28 essential hypertension,29 coronary disease30 and amyloid polyneuropathy.31
In this study, the independent association between the GSTT1 null genotype and CAD (Table 2) was confirmed in the analysis of multilocus epistasis, which may indicate that, at least in this population, the absence of the GSTT1 gene could constitute a marker of genetic risk for the early development of CAD. Previous studies have evaluated the association between the null genotype GSTT1*0 and susceptibility to coronary disease, but the results are inconclusive: some studies found a significant association and others did not.32–36 These inconsistencies can be attributed to the different magnitudes of risk obtained in the different studies, the small effect of the polymorphism on the disease, small sample sizes and the methodology used. From a pathophysiological point of view, the relationship between GSTT1 and atherosclerosis is established enzymatically, through which GSTT1 eliminates toxic metabolites and products of oxidative stress.37,38 In subjects lacking the enzyme, the compounds generated by cell metabolism which are eliminated by the GSTT1 pathway accumulate in the endothelium, leading to endothelial dysfunction.39–41 The second pathway, only recently defined, refers to the role of GSTT1 as a modulator and regulator of proteins which intervene in cell signalling mechanisms.42,43 It has been proposed that GSTT1 sends inhibitory signals to protein kinases responsible for regulating the proliferation of smooth muscle cells in the arterial intima.43 In individuals lacking the enzyme, the proliferation of muscle cells may be exacerbated, and this would contribute to a greater growth of atheromatous plaques compared to subjects carrying the functional enzyme.
There are also discrepancies regarding the association of the MTHFR gene with cardiovascular risk. The majority of studies involved Caucasian and Asian groups, with few reports in Latin populations. A meta-analysis that included 22 age-matched case–control studies found a difference in the average Hcy value between the wild-type CC genotype and the mutated TT (C677T) of 2.7mmol/l.44 The C677T polymorphism and the alteration in folate levels have also been related to DNA hypomethylation and, consequently, to an increased risk of atherosclerosis.45,46 There is evidence that TT genotype carriers have lower genomic DNA methylation values than subjects with wild-type CC.47 However, it has also been reported that not all individuals carrying the thermolabile variant (677TT) have increased levels of Hcy, and that, among those who do, levels return to normal after folate supplementation.48,49 This may therefore be a neutral mutation for Hcy levels when the folate status is adequate.50 Nonetheless, there are few data on the role of Hcy in patients with premature CAD. Hyperhomocysteinaemia has been reported to be an independent risk factor for coronary thrombosis in subjects under the age of 50, rather than for developing coronary artery disease.51,52 There has been very little research in Mexico into the relationship between the MTHFR gene and ischaemic disease of atherosclerotic origin. Assessing 167 patients from Mexico City, Isordia et al. found no independent association between carriers of the TT genotype (C677T) and the risk of premature myocardial infarction (aged ≤45).53 In our study, none of the MTHFR polymorphisms showed independent risk association with early CAD (Table 2). However, based on the results of the multilocus analysis, we do believe that the MTHFR gene, although not enough in itself, is necessary for the development of ischaemic disease, at least in this population. MDR analysis can be useful here as an analytical strategy to investigate the simultaneous effect of different loci and thus detect risk relationships with complex diseases.
With respect to the analysis of epistasis, MDR identified as best model the interaction between the gene GSTT1 and the polymorphism C677T (MTHFR) independent of the variant -108CT (PON1), which may act as an effect modifier in the interaction (Fig. 1). Based on the high- and low-risk genotypes obtained, we can state that GSTT1 and C677T are able to act synergistically: subjects who are carriers of the 677TT genotype (MTHFR) with absence of the GSTT1 gene will have greater susceptibility to developing ischaemic events, while those with the functional GSTT1 gene and the CC677 variant will be more protected. Several studies have reported interaction of GST genes with MTHFR,54–56 but only a few have performed epistasis analyses.57,58 In addition to the mechanisms of epistasis in which a gene's expression can be modified by the action of another or more than one other gene, the GSTT1–MTHFR interaction effect could also occur through epigenetic mechanisms involved in the regulation of gene expression, methylation in particular, alterations of which occur in various diseases. In atherosclerosis, in vitro models have shown that DNA methylation is an early onset process in which both excessive and deficient methylation exists in different genes, while, in the advanced stages of the disease, hyperproliferation of the cells of vascular smooth muscle induces DNA hypomethylation.45,47,59,60
Another possible explanation of the combined GSTT1–MTHFR effect could be the involvement of the enzymes encoded by these genes in the metabolic pathways associated with protection against oxidative stress. Hcy serves as a precursor to the enzymes S-adenosylmethionine (SAM)/S-adenosylhomocysteine (SAH), linked to the methylation and demethylation of DNA, RNA and proteins; and also as a precursor of glutathione (GSH), necessary in the detoxification processes mediated by GST.61,62 It could therefore be said that changes in methylation patterns and alteration in the levels of certain oxidation-reduction metabolism enzymes might be a part of the mechanisms involved in the development of atherosclerosis.
Lastly, gene–environment interaction analysis identified the GSTT1 and MTHFR genes in conjunction with hypertension as the factors that best predict CAD in the study population. As far as the L55M polymorphism is concerned, its presence in the gene–environment model may be due to the linkage disequilibrium with the -108CT variant (LD′=0.394, r2=0.207, p≤0.0001) (Table 4). Previous studies have shown that the pathogenesis of hypertension is associated with alterations in glutathione metabolism.63,64 There is evidence that the mononuclear cells of hypertensive subjects have significantly lower levels of GST than in healthy subjects, and that the activity of enzymes in this metabolic pathway, including GST, also decreases.65,66 These low levels of antioxidant enzymes in hypertension are a result of the oxidative stress conditions prevailing in the vascular environment, and are reversed with antihypertensive treatment.67GSTM1 and GSTT1 null genotypes have also been reported as potential risk factors for the prediction of essential hypertension.67,68
ConclusionsIn this study, epistasis analysis indicates that the combination of different allele variants of antioxidant genes, such as GSTT1 and MTHFR, and hypertension can influence the genetic variability for predisposition to the development of early ischaemic events in the population of Yucatán. MDR analysis can be useful to demonstrate the role of epistatic interactions in the aetiopathogenesis of complex diseases such as CAD.
FundingThis work was funded by clinical-analysis laboratory Biomédicos de Mérida (Yucatán, México).
Conflict of interestsThe authors declare no conflicts of interests.
Please cite this article as: García-González I, López-Díaz RI, Canché-Pech JR, Solís-Cárdenas AJ, Flores-Ocampo JA, Mendoza-Alcocer R, et al. Análisis de epistasia de polimorfismos de genes metabólicos asociados a cardiopatía isquémica en Yucatán. Clin Invest Arterioscler. 2018;30:102–111.