












An alternative route for preparing silicon-substituted hydroxyapatite powders Ca10(PO4)6−x(SiO4)x(OH)2−x (Si-HAp); using different content of SiO4 (0.588–0.48M) in the aqueous solution of Si precursor was investigated. Tetramethylammonium silicate (TMAS) was used as a silicon precursor. The experiments were carried out via conventional hydrothermal treatments at 150°C for 10h. The Si-HAp nanoparticles obtained were characterized by X-ray diffraction (XRD), Fourier transform IR spectroscopy (FTIR), inductively coupled plasma AES (ICPAES) and scanning electron microscopy (SEM) techniques. A drastically decrease of the diffraction was not observed by increasing the Si content in the structure of the hydrothermally Si-Hap particles produced. A detailed structural analysis conducted by Rietveld refinement indicated that the incorporation of Si occurred in hydroxyapatite (HAp) lattice by partially replacing phosphate (PO43−) groups with silicate (SiO44−) groups. Raman and X-ray photoelectron spectroscopy (XPS) analysis confirmed the substitution of PO43− by SiO44− in the Si-HAp structure. The crystallite size decreases by increasing the Si molar content in the hexagonal HAp structure. TEM observations revealed that the Si-HAp produced have a high crystallinity. A slight decrease of the particle size of the Si-HAp compared to pure HAp was observed (from 60–100nm to 40–70nm). The morphological analysis revealed that all the SiHAp powders prepared have the rod-like shape. Additionally, a slight decrease in both length and diameter of nanorods was observed by increasing the silicon content, the minimum size obtained for the powders containing the largest amount of Si (20Si-HAp) 25 and 70nm. The presence of Si in the structure of the Si-HAp favored densification of the compacts treated at high temperature 1200°C in comparison with pure HAp.
Una ruta alternativa de síntesis de polvos de hidroxiapatita (HAp) sustituida con silicio Ca10(PO4)6−x(SiO4)x(OH)2−x, (Si-HAp) utilizando diferentes concentraciones molares de SiO4 (0.588-0.48M) en la solución precursora de silicio (Si). Se utilizó solución de silicato de tetrametilamonio (TMAS, por sus siglas en inglés). Los experimentos se realizaron mediante tratamiento hidrotérmico convencional a 150°C y 10h. Las nano partículas de Si-HAp obtenidas se caracterizaron por difracción de rayos X (XRD), espectroscopia de IR con transformadas de Fourier (FT-IR), análisis por espectrometría de plasma acoplado inductivamente (ICP-AES) y microscopía electrónica de barrido (SEM). El incremento del contenido de Si no cambió drásticamente la intensidad de los picos de difracción. El análisis estructural por refinamiento Rietveld indicó que la incorporación de Si efectivamente ocurrió en la red de HAp reemplazando parcialmente los grupos fosfato (PO43−) con grupos de silicato (SiO44−). Análisis por espectroscopia Raman y (XPS) confirman la sustitución de PO43− por SiO44− en la estructura Si-HAp. El tamaño del cristalito disminuye con el aumento del contenido de Si en la estructura hexagonal de HAp. De las observaciones de TEM, confirman la alta cristalinidad del polvo producido. Se observó una ligera disminución del tamaño de partícula de Si-HAp en comparación con la HAp pura (de 60-100 a 40-70nm). La morfología de las muestras Si-HAp reveló la obtención nanopartículas de tipo varilla. Se observó una ligera disminución en la longitud y diámetro de los nano varillas con el aumento del contenido de Si, con un diámetro de 25-45nm y una longitud de 70-90nm para el 20SiHAp. La presencia de Si en la estructura de la Si-HAp favoreció la densificación de los compactos, particularmente para los obtenidos a altas temperatura de 1200°C en comparación con la HAp pura.
Many researchers have recently conducted significant efforts to study the synthesis of silicon-substituted hydroxyapatite (hereafter referred as Si-HAp) toward for improving the bioactivity [1,2]. In general, with a vast improvement and others with some disadvantage, a reduced number of research works focused to synthesize Si-HAp particles are based on the titration of calcium hydroxide [3] and sol–gel [4]. The precipitation method using calcium nitrate and TEOS as a precursor of silicon produces a Si-HAp powder with a considerable content of by-products, namely TCP; or other phosphates which are formed in the sintering stage. A wet chemical method approach had also been proposed to produce Si-HAp particles, but the crystal growth of the particles promoted during the precipitation stage produces large Si-HAp crystals. From this result, they indicate that SiO44− ion substituted PO43− group site in apatite structure. Also, they found that appropriated amount of TEOS doping ratio was 10 Mass %, for highest biocompatibility behavior, because of the formation of SiO2 aggregation in primary content than 15% weight of TEOS [5]. Some others, report that the crystallite size and fraction of the crystalline phase decrease with the increase of silicon content [6–8]. The hydrothermal technique typically promotes the formation of materials with a high degree of crystallinity and with a stoichiometric Ca/P ratio of hydroxyapatite. Kim et al. [7] reported that porous silicon incorporated on hydroxyapatite was prepared by hydrothermal treatment and solvothermal treatment of natural coral repeatedly. Typically, the hydrothermal technique enhances various advantages such as the formation of single-phase particles with a homogeneous monomodal particle size distribution, and a high degree of crystallinity [8,9]. The preparation of hydroxyapatite nanoparticles is important because of the broad number of applications in various industries, in the biomedical field and for preparing dental devices; which includes toothpastes and cements products [10–12]. Moreover, it is known that the tetramethylammonium ion (C4H12N+) contributes to the formation of double four membered cage silicate ions [11,14]. According to the literature review, in the present work, we aimed to study the feasibility for producing Si-substituted HAp particles using an alternative silicon precursor that triggers the incorporation of silicon in the HAp structure in an aqueous solution system under hydrothermal reaction conditions; and we have attempted to control the particle size coarsening, as well as silicon content. The usage of the tetramethylammonium silicate (TMAS) precursor is likely to limit the formation of reaction by-products during the synthesis by hydrothermal process. The TMAS is an adequate silicon precursor due to its chemical composition that might provoke the control of the hydrothermal media pH because produces the sufficient content of ammonium ions, and the presence of these four ions makes it more soluble in comparison with the other related specie such as tetraethyl orthosilicate TEOS [12].
Experimental procedureStoichiometric hydroxyapatite (HAp) and the partially Si-substituted hydroxyapatite (Si-HAp) powders were synthesized by the conventional hydrothermal method. All the chemicals employed were reagent grade (Sigma Aldrich, 99.99% purity). Any purification of all chemical reagent precursor was conducted prior the solutions preparation, a 0.6M solution of tetramethylammonium silicate ((CH3)4N(OH)·2H2O), TMAS; number CA#24867466 and Aldrich 438669) was prepared by diluting with deionized water the stoichiometric volume of the TMAS solution (TMAS average content of 20.0wt.% in water). The solutions of Ca(NO3)2·4H2O (1M) and (NH4)2HPO4 (0.6M) were prepared by weighing the stoichiometric content of both salts, and then were dissolved separately in distilled water. The volume of each precursor was added according to the stoichiometric molar ratio established for the partially substituted Si-HAp, Ca/P+Si=1.67. Hence, the volume molar mixture that corresponds to Ca:P:Si was 10:6:0 (HAp), 10:5.76:0.24 (4Si-HAp), 10:5.4:0.6 (10Si-HAp), 10:5.04:0.96 (16Si-HAp), 10:4.8:1.2 (20 Si-HAp). The solutions of TMAS and PO43− were initially mixed under stirring, then was added the volume molar of the Ca2+ precursor to the mother liquor, the addition of this solution produced the formation of a colloid. The total volume of the three precursor solutions mixed was of 50ml, which corresponds to the 70% filling ratio of the micro autoclave Teflon vessel. The pH of all the mixed colloidal suspensions was adjusted to a value of 10.00±0.1 by adding a certain volume of a NH4OH solution dropwise under agitation [15,16].
The colloidal suspension was transferred to Teflon vessels of the autoclave and heated at 150°C, using a convection furnace during 10h. After the reaction finish, the powders were washed several times with deionized water up to neutral pH. The powder was dry by using freeze drier to avoid aggregations of the particles. Subsequently, 0.25±0.005g of HAp and Si-HAp powders were compacted using a load of 60MPa for 5min, the compacts then were sintering at different temperatures. The characterization of the microstructure of the HAp and Si-HAp particles was conducted using TEM field emission (FEI TITAN Phillips 80–300kV microscope) operated at the accelerating voltage of 300kV. TEM images and electron diffraction patterns (SAED) were obtained using a Gatan CCD digital camera with 1024×1024 pixels and resolution of 1k. The optical conditions of the microscope were as follows: spherical aberration Cs=1.25mm, 1Å limit, point–point resolution of 1.4Å, α=8.4mrad. X-ray spectra were simultaneously obtained using scanning transmission electron microscopy (STEM) imaging, producing scan lines, areas (chemical mapping), and specific chemical analysis. The optical conditions at which the STEM images were obtained were as follows: STEM-EDS nano Probe operated at 300kV, extraction voltage 4500V, gun lens 3, spot size 6, and C2=70μm. And image analysis was carried out using an Image-Pro® Plus software.
The crystalline phases and the changes on lattice parameters associated with Si incorporation into the hexagonal structure of HAp phase and Si-HAp were determined by X-ray diffraction analysis. Diffraction patterns were collected in a range 2θ from 15 to 130° at a scanning speed of 1°/min and a step size 0.002° in a 2θ/θ scanning mode using an X-ray diffractometer (Rigaku Ultima IV, Japan) equipped with Cu Kα radiation (α=1.54056Å) at 40kV and 20mA. Furthermore, Rietveld refinement of samples containing single-phase HAp and Si-HAp reaction product were carried out to determine the crystallite size and lattice parameters using the TOPAS software (TOPAS, version 4.2; Bruker AXS: Karlsruhe, Germany, 2009). The background was modeled by a shifted Chebyshev 6-coefficients polynomial function while the pseudo-Voigt function was selected to fit the profile peak shape. FT-IR analyses were conducted in an FT-IR JASCO 4000 spectrometer to determine aspects related the variations in the vibrational modes of OH− and PO43− functional groups. The FT-IR analyses were carried out using palletized samples consisting of a mixture of 5mg of HAp or Si-HAp and 200mg of KBr. The samples were dried in an oven for 12h at 100°C in air before the pellet preparation. FT-IR spectra were run at a wavelength range of 400–4000cm−1 in the transmittance mode. The contents of Ca, Si and P in the reaction products were quantified by the inductively coupled spectrometer (Shimadzu, ICP Spectrometer ICP-9000). Additionally, the morphology and particle size distribution were examined by scanning electron microscopy (FE-SEM JEOL 6500F, Japan), previously of the observations, all the samples were sputter-count with Pt, to avoid charging effects. In addition, the apparent densities of the compact specimens of HAp and Si-HAp were determined based on Archimedes’ principle using a helium pycnometer (Multipycnometer Quantachrome). Prior to the density measurements, the pycnometer was calibrated using a standard stainless-steel sphere with a volume of 56.5592cm3, which was located inside the measuring cell (volume 135cm3). The foamed specimens were previously weighted, and the volume was measured three times at a helium pressure of 0.117MPa. The chemical composition of the SiHAp powder as prepared samples was determined by X-ray photoelectron spectroscopy (XPS). XPS spectra were recorded on a Kratos spectrometer (Japan) operated using Al Kα (1486.6eV) monochromatic X-ray source. The XPS study was carried out in the VersaProbe II equipment of the company PHI, with a vacuum in the analysis chamber of 2×10−8mTorr, a monochromatic X-ray source with aluminum anode and a radiation energy of 1486.6eV was used. The general spectra (survey) were obtained with a step energy of 117.4eV, the analysis region was 1400–0 (eV) in link energy. High resolution spectra of the C 1s, Ca 2p, P 2p and O 1s signals were obtained for each of the samples. The high resolution spectra were acquired with a step energy of 11.75eV. The deconvolution of these spectra was performed by adjusting Gaussian curves, leaving their position and area without restriction, however the FWHM value remained fixed in each of the adjusted curves. In addition, Raman characterization was carried out in a Horiba Jobin team in regions of 200–4000cm−1, using a grid of 600T. The excitation source was formed by a laser of 532nm with a 25% filter, goal of 100; the time of obtaining data for this analysis was 120s.
Results and discussionSynthesis of Si-HAp powders and its structural characterizationThe chemical composition of the HAp and Si-HAp powders prepared under conventional hydrothermal treatment determined by ICP-AES. Table 1 shows the results including the calculate (Ca/P) and (Ca/P+Si) ratios and lattice parameter. It is noteworthy that in all runs experiment under the hydrothermal conditions, the increase of SiO44− solution concentrations promoted the optimization of the incorporation of Si in the structure of the HAp during the hydrothermal treatment. The progressive saturation of the hydrothermal medium with Si ions provoked a sufficient control of the chemical composition in the Si-HAp crystallized, the degree of Si incorporation was near to 61mol% in the powders regardless the nominal content added in the reactor. This behavior was remarkable for the particles prepared with a large amount of silicon, 20mol% Si (20Si-HAp). The deficiency on the Si incorporation in the hexagonal HAp structure is it might be enhanced due to the formation of a large amount of the intermedium species by the addition of TMAS, such as SiO44− [Si(OSi)3(O−)], N(CH3)4OH·5H2O and C4H13NO·3H2O. These species a chemically stable in aqueous solution [11], which avoids the incorporation of the total nominal Si content added to the reaction system during the crystallization step, the concentration of the compound in the medium increased until the selected stoichiometric value is obtained. To provide evidence for these results, wet chemical analyses of the remaining mother liquor were conducted, aiming to elucidate the above inference, the analyses were conducted on those remaining mother liquor corresponding to those experiments aimed to prepare the solid-solutions supersaturated with Si4+ ions. The ICP confirmed the presence of small amount of unreacted Si and no traces of Ca and PO43− in the remaining mother solution after hydrothermal treatment. The ICP analysis of Si ions conducted for the remained mother liquor after the reaction indicated that the average content of this element was 0.02±0.001mol/ml. It is worth mentioning that the real content of the Si determined by chemical analysis for the prepared powder corresponds only near to the 61% regardless to the nominal the content selected for the Si precursor (TMAS).
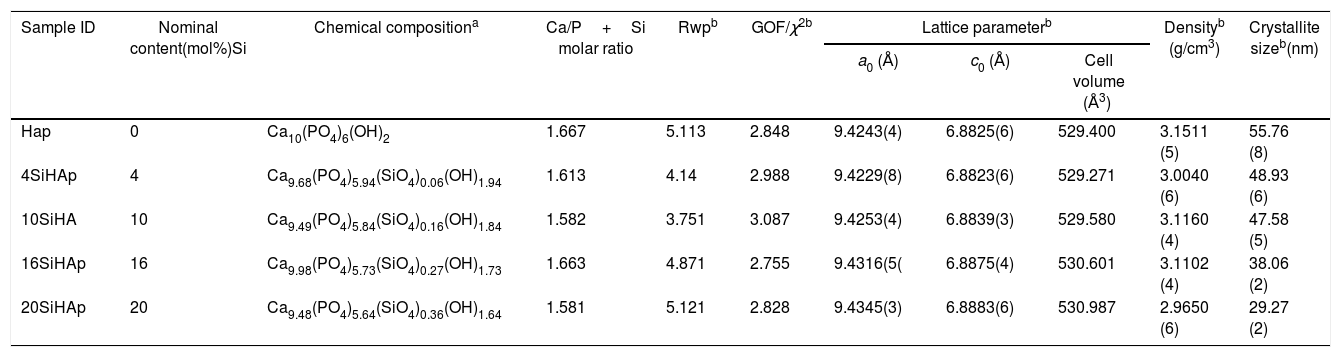
Chemical and physical features of Si-HAp fine particles prepared with different Si4+ concentration by hydrothermal synthesis at 150°C for 10h.
| Sample ID | Nominal content(mol%)Si | Chemical compositiona | Ca/P+Si molar ratio | Rwpb | GOF/χ2b | Lattice parameterb | Densityb (g/cm3) | Crystallite sizeb(nm) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a0 (Å) | c0 (Å) | Cell volume (Å3) | ||||||||
| Hap | 0 | Ca10(PO4)6(OH)2 | 1.667 | 5.113 | 2.848 | 9.4243(4) | 6.8825(6) | 529.400 | 3.1511 (5) | 55.76 (8) |
| 4SiHAp | 4 | Ca9.68(PO4)5.94(SiO4)0.06(OH)1.94 | 1.613 | 4.14 | 2.988 | 9.4229(8) | 6.8823(6) | 529.271 | 3.0040 (6) | 48.93 (6) |
| 10SiHA | 10 | Ca9.49(PO4)5.84(SiO4)0.16(OH)1.84 | 1.582 | 3.751 | 3.087 | 9.4253(4) | 6.8839(3) | 529.580 | 3.1160 (4) | 47.58 (5) |
| 16SiHAp | 16 | Ca9.98(PO4)5.73(SiO4)0.27(OH)1.73 | 1.663 | 4.871 | 2.755 | 9.4316(5( | 6.8875(4) | 530.601 | 3.1102 (4) | 38.06 (2) |
| 20SiHAp | 20 | Ca9.48(PO4)5.64(SiO4)0.36(OH)1.64 | 1.581 | 5.121 | 2.828 | 9.4345(3) | 6.8883(6) | 530.987 | 2.9650 (6) | 29.27 (2) |
Moreover, the slight deficiency in calcium can be due to instrumental and weigh error during the preparation of calcium nitrate tetrahydrate solution. The substitution mechanism of silicon to HAp would describe the substitution of a phosphate group for a silicate group, with an appropriate rearrange for charge balance, is given in Eq. (1): to compensate for the extra negative charge of the silicate groups, some of the OH− groups would be lost to retain charge balance.
Typical XRD patterns and Rietveld refinement plots of the HAp and Si-HAp species prepared under hydrothermal conditions at 150°C, 10h with different content of mol% Si, using [(CH3)4N(OH)·2SiO2] (TMAS) as Si precursor as it is an exhibit in Fig. 1. All diffraction patterns of samples of silicon-substituted hydroxyapatite can be indexed based on the card, JCPDS No. 09-0432, with the hexagonal structure of the HAp, without the presence of any other phase related to the formation of silicon oxides or sub-species of calcium phosphate. On the diffraction patterns, the increase of Si content (Fig. 1) did not seem to affect the diffraction pattern of hydroxyapatite drastically. However, it promotes a slightly decreased on the peak intensity, which it is consistent with the results elsewhere reported when the TEOS was used as precursor [14]. Furthermore, a clear displacement to lower 2θ angles, and a diffraction peak broadening resulted from increasing the Si content incorporated into the HAp structure (Fig. 1) in our specimens, in particular for those prepared with Si content over 10mol%. This result was disagreeing to the previously reported for other authors when they used TEOS as Si precursor [7,15]. Nevertheless, using the current tetramethylammonium silicate (TMAS) as a precursor of Si in aqueous solution, it was evident that an increased of Si content did not change drastically the crystallinity, but the size of the crystallite was decreasing significantly as it can be seen in Fig. 1, as well as is indicated in Table 1.
, which were prepared under hydrothermal conditions at 150 ̊C for 10 h.")
The Rietveld patterns support these results as it is shown in Fig. 1(b) which were calculated considering that the total content of Si incorporated in the HAp structure was at PO43− sites, as suggested elsewhere [6,12,17]. The selected refinement approach provided the lowest values of the goodness-of-fit factor (GOF or χ2), which were below 2.8 (Table 1), indicating the excellent accuracy of the refinement conducted in this case.
Also, the partial substitution of the PO43− group with SiO44+ provoked a slightly decrease in Si-HAp lattice parameters up to a Si content higher than 4mol%. However, an increase in “a0” and “c0” occurred for the silicon hydroxyapatite samples prepared with 10mol% of Si. This phenomenon might be associated with variations in the bond length of OH− within the Si-HAp structure. The Rietveld analyses indicated that the incorporation of Si up to 20mol% promote a slight and progressive decrease of the crystallite size and steadily increase the cell volume as is shown in Table 1.
The present results are in agreement with those previously reported by Gibson et al. [3], whom had found that the silicon substitution in the HAp lattice resulted in an abnormal behavior of the hexagonal structure lattice parameters; which correspond to decrease of the “a” axis, while an increase in the “c” axis usually occurs. The partial Si substitution in the PO43− sites of the hexagonal structure, also caused a decrease in the number of hydroxyl (OH−) groups in the unit cell, which was expected from the proposed substitution mechanism and is required for the total balance charge. Additionally, the Si incorporation in the HAp lattice resulted provoked a gradual distortion of the PO4 tetrahedra, which is indicated by the increase in the distortion index. These inferences are in a good agreement with our results obtained by the structural FT-IR and Raman analyses and the chemical compositional XPS analysis as well, which are discussed in detail in the next section.
These present results agreed with those reported elsewhere [12,3] when the precursor of silicon was TEOS, because with that precursor usually the crystallite size decrease with the incorporation of Si. Although, the small differences were found in this investigation con respect to use TEOS, a systematic decrease of OH− with the increase Si content during the reaction because the formation of intermedium species of Si as it was mention above, among the solubility of the Si species in water under hydrothermal conditions could contribute to the rapid consume the OH−, and this inference still not elucidated yet. Despite of this lack of information, we surmise that this phenomenon might be attributed to variations in the bond length of OH− within the HA structure. The results of the FT-IR analyses support this inference, Fig. 2, which revealed a systematic reduction in the vibrational OH− band at 3571cm−1 with increasing the Si content in the HAp structure.
![FT-IR analysis of the Si-HAp powders obtained by conventional hydrothermal treatment at 150°C, 10h with different Si content using [(CH3)4N(OH)·2SiO2] (TMAS) as a Si precursor.](https://static.elsevier.es/multimedia/03663175/0000005900000002/v1_202004071154/S0366317519300512/v1_202004071154/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd3TCEZuNxhudow/+3Hrh9WJc+mtnYSSPL9K6mfMWVj1nTuSYcv/6N3dvYkGNuXY7n5sUpJZGkWRubFooWFg3+POJZhI4+yxpWssMb78dDtWQnvvwgNC8KX+MJyZatBhwUJvxAq6xUXwy+YRNu6kOQFKSs0i9QFHqHt3os7vkgvX/KAKN2XcmTv3Ie8frJdzwwz9ZuVEZJMdDnzBLZuNNAOhifo13IzXwG3wn49whyS3UvYYEiQcUfswAIiCE3TV2Q= "FT-IR analysis of the Si-HAp powders obtained by conventional hydrothermal treatment at 150°C, 10h with different Si content using [(CH3)4N(OH)·2SiO2] (TMAS) as a Si precursor.")
In Fig. 2 the typical bands at wavelengths of 3571 and 631cm−1 revealed in the Si-HAp powders hydrothermally produced, which corresponds to the stretching and vibration modes of the hydroxyl (OH−) groups; these bands are observed in the both HAp and Si-HAp powders. The large bands at 1084, 1037 and 954cm−1 that belong to PO group stretching vibration modes, while the doublet at 593–572cm−1 corresponds to the bending mode of the OPO bond, bending to match the data reported in the literature and are associated with HAp [8]. Noteworthy, the signals of the ν1 and ν3 bending mode of POP steadily decrease as the increase the Si content, and simultaneously the symmetry of the PO boned progressively change, which it is attributed to the incorporation of the SiO44− ions, as well as a very small, displace to the lower wave number of the bands. The FT-IR spectra showed a significant difference with the addition of different content of silicon precursor (Fig. 2). In comparison with the FT-IR spectra of the pure HAp sample, the intensity of the peak at 1084cm−1 of the Si-HAp samples decrease as the silicon content uptake increases, this process caused the crystal structure change of the HAp lattice induced by Si doping [16,17]. The presence of the SiO44− bands was revealed at wavelength of 970cm−1, and this band is visible on those samples prepared with high molar contents over 10mol% Si.
According to the present results, the FT-IR analyses of the Si-HAp, these indicated that the amount of silicon incorporated into the HA lattice was smaller than that selected in each case. However, the silicon substitution appeared to affect the FT-IR spectra of the HAp, in particular the vibrational bands associated to the PO group. The significant effect of the silicon addition in the particles produced hydrothermally, is depicted by the marked the decrease in the hydroxyl stretching bands at 3571cm−1 and 631cm−1 and phosphate stretching bands at 965cm−1. According with this result, we surmise that these structural variation might be related with the uptake of SiO44− tetrahedrons that substitute the PO43− units in the hydroxyapatite structure. Under hydrothermal conditions the incorporation of silicon ion in the hydroxyapatite phase induces a reduction of the amount of hydroxyl groups to compensate for the extra negative electric charge produced by the incorporation of the silicate groups, and the OH− group vacancies formation is likely to proceeded to maintain the charge balance, as is depicted by the following equation PO43−+OH−→SiO44−+V(OH). Indeed, this behavior is related to the substitution of PO43− by SiO44− tetrahedrons revealed by the FT-IR analyses which is consistent with the previous report by Gibson et al. [3]. Furthermore, the results were confirming by Raman spectroscopy analysis of the synthesized samples of SiHAp.
Fig. 3 revealed that the four vibration modes of phosphate tetrahedra are observed in the spectra range of 200–1300cm−1. The intense ν1 mode is indicated by the band at 960cm−1 corresponding to the symmetric stretching of PO bonds, the ν2 modes around 425cm−1 corresponds to doubly degenerate OPO bending modes, the ν3 modes around 1042cm−1 and ν4 around 600cm−1. Furthermore, on the central part of the spectra, Fig. 3 shows the detail for the ν1 mode, where the PO4 symmetric stretching mode. The mainly signal around 955cm−1 corresponds to HAp structure, main phase of the samples. A new band appeared for the Si-HAp samples, which is more visible for the sample with Si contents over 4mol%, at 888cm−1. This signal was gradually increasing as the incorporated silicon content increased. In contrast, the OH− signal was gradually decreasing with a broadening of the band as was mentioned elsewhere [20]. In addition, the almost absence of the OH− vibration band for the sample synthesized with content of 16 and 20mol% of Si indicates the modification of the hydroxyl when the silicate was substituted. This result might the reason that the crystallite increases the size more in axis “a”. This result is in agreement with the XPS analysis, which was conducted to confirm the presence of Si in those Si-HAp particles hydrothermally prepared.

Fig. 4 shows the typical XPS spectra which were conducted to investigate the substitution of PO43− ions with SiO44− ions in the hexagonal HAp structure. Generally, the spectra of the synthesized HAP and Si-HAp powders with different contents of Si are shown in Fig. 4(a), XPS observation indicates that the chemical elements Ca, P, O and C were present in the samples, and the Si signals were detected on the prepared Si-HAp samples, the variation on the intensity of the Si peak is due to the increase of the silicon content. Fig. 4(b)–(g) shows the XPS spectra of the C 1s, O 1s, Ca 2p, P 2p and Si 2p signals for the samples of HAp and Si-HAp respectively. In Fig. 4(b) all the peaks for the C 1s are asymmetric with the faint shoulder at high binding energy, which is consistent with the reported by Shing et al. [21]. In addition, the intensity of the signals decreases as que silicon content increases. The C 1s was deconvoluted to three peaks which corresponded to aliphatic carbon atoms (284.5±0.2eV). The detection of carbon should be due to the absorption of CO2 gas from air during the preparation determination.
 HAp and Si-HAp powders prepared under hydrothermal conditions at 150 °C, during 10 h using TMAS as Si precursor. And (b-g) binding energy curves (eV) of Ca 2p, O 1s, Si 2p and C1s of each powder samples in Fig 4a.")
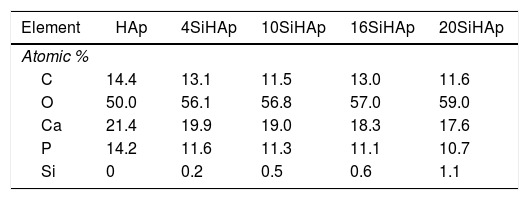
On the other hand, Ca 2p XPS signals can be observed in the spectrum portrayed in Fig. 4(c), which correspond to the doublet that correspond to CaO bonds. The Ca 2p spectrum showed a doublet with Ca 2p3/2 at 347.0±0.3eV and Ca 2p1/3 at 350.55±0.2eV. The samples prepared with 4 and 10mol% of Si content shows a small change, mainly a decrease of the intensity and slight decreased of the bending energy of Ca 2p3/2 at 346.83±0.3 and 346.87eV. This decrease can explain the deficiency on calcium of the powder synthesized with high content of Si and is consistent with ICP analysis and reported by Rietveld analysis of Table 1. In contrast, the powders with Si contents of 16 and 20mol% observed a slightly increase on the binding energies 347.66 and 347.5eV; respectively. Furthermore, the P 2p peak was symmetric as is revealed in Fig. 4(d) indicating that its binding energy was quite stable in apatite. It was referenced at 133.15; 133.0; 132.7; 132.50 and 133.4eV for the samples prepared with 0, 4, 10, 16 and 20mol% Si, respectively. However, a small decrease the intensity was evident and for samples with 16 and 20mol% of Si and a small displacement to the high bending energy. The O 1s peak showed an asymmetry and was fitted using the peak deconvolution approach. It was decomposed into three components (Fig. 4(e)). The main component at 531.2±0.2eV corresponded to the O linked only to a phosphorous atom as in PO43− ions. The second at higher energy (532.58eV) could be attributed to SiO peak, and the binding energy slightly decrease for the samples synthesized with 4, 10 and 16mol% of Si. Fig. 4(f) and (g) shows that the Si 2p spectrum has decomposed into 103.6±0.3 and 102.4±0.3eV, with an area intensity ratio of about 2:1 for 20 Si-HAp powder. In contrast, for the 10Si-HAp samples the Si 2p spectrum ratio was 1:2 this inference is attributed to the gradual formation of SiOSi bond substitution of Si in the HAp. As it was reported elsewhere [18], we found that the binding energy of SiO44− in silicate was centered at 102.4±0.2eV for powders 4Si-HAp and 10Si-HAp respectively. According to the peak of SiO2 was centered at 103.9eV [22], the first component for the samples with 10Si-HAp and 20Si-HAp was assigned to Si-O2 that can be attributed to silicon atoms becoming to isolated SiO44−. Furthermore, from this XPS analysis was possible to confirm the influence of adding different %mol of Si in the HAP structure during the hydrothermal treatment. The small changes on the atomic composition of the O, P, Ca and Si on the HAp and Si-HAp structure can be seen in Table 2. It was found that the increase of %mol of Si in the HAp produces a slightly decrease of the % atomic of Ca and P, but promote an increase of atomic content of O and Si, that confirm clearly the substitution of the SiO44− by PO43−. From Raman and XPS analysis we conclusively can demonstrate that a large amount (near 61% nominal) of the added SiO44− was possible to introduce into the HAp structure the SiHAp.
Morphological evaluation of the hydrothermally prepared HAp and Si-HAp powdersFE-SEM micrographs of the hydroxyapatite powders substituted with different molar % of Si are shown in Fig. 5(a)–(d). Those results show a small decrease in particle size, as well as, small changes on morphology were note with the increase of Si in the samples of Si-HAp compared to pure HAp powder. This behavior is consistent with the results of X-ray diffraction patterns and the lattices parameter after Rietveld analyses (Fig. 1b and Table 1). It deserves emphasizing that the addition of Si ion promotes a small but significant crystalline and size reduction for the powder of the prepared samples with the large amount of SiO44− incorporated into the HAp structure, confirming the results reported elsewhere [7]. Also, a systematic increase of Si content has a notable influence on the morphology of the particles. Samples prepared with low content of Si shows a spherical and regular morphology homogeneous. With the increases in the molar concentration of Si, the obtained particles appear as elongated grains (needles type). The observed small crystal size of Si-HAp can be explained regarding higher nucleation density during hydrothermal precipitation process as was mention previously [16–18]. TEM images of the prepared Si-HAp powders under hydrothermal conditions at 150°C, 10h are shown in Fig. 6(a) HAP, 6(b) 10Si-HAp and 6(c) 20Si-HAp respectively. The primary size of the nanorods observed corresponded to be between 50–100±2.5nm for the powder prepared with 4mol%.
 0, (b) 4, (c) 10% and (d) 20mol% of Si4+.")
![TEM micrographs of silicon hydroxyapatite (Si-HAp) particles prepared under hydrothermal conditions at 150 ̊C, 10 h with (a) 4 (b) 10 and (c) 20% molar of Si+4 using [CH3)4N(OH)•2SiO2] (TMAS). and particle size distribution of (d) HAp, (e) Si-HAp 4 and (f) 16 mol % of Si.](https://static.elsevier.es/multimedia/03663175/0000005900000002/v1_202004071154/S0366317519300512/v1_202004071154/en/main.assets/gr6.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd3TCEZuNxhudow/+3Hrh9WJc+mtnYSSPL9K6mfMWVj1nTuSYcv/6N3dvYkGNuXY7n5sUpJZGkWRubFooWFg3+POJZhI4+yxpWssMb78dDtWQnvvwgNC8KX+MJyZatBhwUJvxAq6xUXwy+YRNu6kOQFKSs0i9QFHqHt3os7vkgvX/KAKN2XcmTv3Ie8frJdzwwz9ZuVEZJMdDnzBLZuNNAOhifo13IzXwG3wn49whyS3UvYYEiQcUfswAIiCE3TV2Q= "TEM micrographs of silicon hydroxyapatite (Si-HAp) particles prepared under hydrothermal conditions at 150 ̊C, 10 h with (a) 4 (b) 10 and (c) 20% molar of Si+4 using [CH3)4N(OH)•2SiO2] (TMAS). and particle size distribution of (d) HAp, (e) Si-HAp 4 and (f) 16 mol % of Si.")
Although the powders prepared with 10mol% of Si Fig. 6(b) consist of nanorods with an elongated grain size in the range between 40 and 90nm and the thickness between 25–45±2.8nm respectively, and the nanorods for the as indicated from the size distributions, Fig. 6(d)–(f). The mean particle size determined from TEM images was similar to that the value calculated by the Rietveld refinement (Table 1).
The grain size of the Si-HAp samples was smaller than that of pure HAp, and this behavior it was not evident for the powders of Si-HAp prepared with content Si 4 or 16mol%. Until now, as was mentioned by Gibson there are many reports relative to the synthesis of Si-HAp emphasizing the decreased of the grain size with the increase of Si, and some authors refer to have a limit of 0.5% of Si, over this content, the decreasing of grain is very slow [17]. In this case, the decrease of the grain size was also slightly, and it can be associating to the hydrothermal conditions and chemical precursor used for this synthesis as well as the % mol of Si content, it will still need more detail investigation using the TMAS precursor.
Microstructural and structural aspects of the sintering HAp and SiHAp compactsSintering experiments were carried out to verify the changes on the microstructural and structural behavior of the prepared HAp, and Si-HAp. The experiments were run using the prepared powder under hydrothermal conditions at 150°C and 10h, consequently, the powders were compacted using a load of 60MPa for 5min and subsequently heat treatment at various temperatures. In general, pure HAp and Si-HAp showed smaller particle size at this temperature and the relative density of the sintered HAp corresponds to be 3.175±0.04g/cm3, with increase of the Si % mol the apparent density slightly decreased to 3.037, 3.150, 2.926 and 3.150±0.04g/cm3, for samples with 4, 10, 16 and 20mol% of Si, respectively. Micrographs of the surface of pellets sintered at 1200°C are shown in Fig. 7(a)–(d) for the prepared samples with (a) 0, 4, 10, 16 and 20mol% of Si respectively. The morphology of the sintering compacts is mainly consisting of nano-sized spheroidal shaped crystallite. As the heating temperature increased to 1200°C the size of the grain was largest in comparison with the SiHAp samples (Fig. 7(a) and (b)). Besides, 4Si-HAp and 16Si-HAp samples show a nanostructure similar to that of the HAp sample, however residual nanoporosity remains on these samples and as it was observed on the micrograph (Fig. 7(b)–(d)) for the compacts prepared with 4, 10 and 16% of Si content of Si-HAp, that corroborate the slightly decrease of the density (Table 2).
 HAp and Si-HAp samples containing (b) 4, (c) 10 (d) 16 and (e) 20 mol %. The elemental mapping conducted by EDX of the samples produced with (f) 16 and (g) 20 mol % Si are also included.")
It is worth mentioning that the compacts prepared with higher content of silicon at 1200°C show some small bright particles that were formed during the sintering process which can be attributed to a dissolution of the excess of silicon at high temperatures. Also, the smaller grains and more homogeneous microstructure for HAp and low content of Si-HAp specimens, led to denser sintered bodies at the selected sintering temperature. Microanalysis (EDX) and high resolution mapping on the samples prepared with high content of Si (16 and 20mol%) are confirming the presence of the Si in the grains, and the formation of small particles of the SiO2 as a segregates, which are consistent with the EDX (Fig. 7(e) and (f)) and Rietveld analysis (Table 1) as above-mentioned.
In addition, Fig. 8(a) and (b) shows the XRD patterns of the sintered compacts at different temperatures, thermal decomposition was detected for both powders of the prepared HAp and Si-HAp. At a temperature lower than 1000°C, a small displacement of the peak to smaller angles 2θ (210) which was assigned to the formation of a new phase of β-TCP (JCPDS-09-0169), at higher temperature over 1100°C, the β-TCP phase disappears altogether. Also, for the HAp powders at a temperature of 600°C, the formation of a small peak of α-TCP (JCPDS-09-0348) phases appears as it can see on XRD diffraction patterns in Fig. 8(a). On the other hand, the transformation of the HAp phases to α-TCP phases on the Si-HAp was more evident; this behavior can be attributed to the presence of Si on the HAp structure. Also, the steady increase in the intensity of the peaks increases as Si content increase also depends on the temperature of heating as was mentioned elsewhere [17–19]. Those results are consistent with XRD patterns and SEM observations. Figure 9 shows the FT-IR spectroscopy analysis of the Si-HAp sintered compacts at 1200°C, small signal of the hydroxyl stretching band appears at 3573.28cm−1, for the Si-HAp sintered compacts, the presence of the SiO44− bands can be detected at wavelengths 802.11cm−1 and 802.96cm−1 and those bands were evident for the powder of 16Si-HAp and 20Si-HAp respectively. Also, small new bands relate also related with the presence of SiO44− bands with the SiOSi were detected for the samples 10Si-HAp 16Si-HAp and 20 Si-HAp at wavenumber 725.56 and 731.86cm−1. Those results are agreeing with the SEM observation for the samples with 16 and 20mol% of Si that form small segregates of SiO2 in the grain limit and explain why the silicon content on the samples prepared with 16 and 20mol% was low in comparison with the initial concentration of added on the Si precursor.
 HAp (600–1300°C) and (b) 16Si-HAp (400–1200°C) respectively.")
![FT-IR analysis of the sintered Si-HAp powders at 1200°C with different Si content (0–20mol%) using [(CH3)4N(OH)·2SiO2] TMAS as a Si precursor.](https://static.elsevier.es/multimedia/03663175/0000005900000002/v1_202004071154/S0366317519300512/v1_202004071154/en/main.assets/gr9.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd3TCEZuNxhudow/+3Hrh9WJc+mtnYSSPL9K6mfMWVj1nTuSYcv/6N3dvYkGNuXY7n5sUpJZGkWRubFooWFg3+POJZhI4+yxpWssMb78dDtWQnvvwgNC8KX+MJyZatBhwUJvxAq6xUXwy+YRNu6kOQFKSs0i9QFHqHt3os7vkgvX/KAKN2XcmTv3Ie8frJdzwwz9ZuVEZJMdDnzBLZuNNAOhifo13IzXwG3wn49whyS3UvYYEiQcUfswAIiCE3TV2Q= "FT-IR analysis of the sintered Si-HAp powders at 1200°C with different Si content (0–20mol%) using [(CH3)4N(OH)·2SiO2] TMAS as a Si precursor.")
Si-HAp powders were successfully obtained under conventional hydrothermal synthesis conditions at low temperature 150°C for 10h using TMAS as Si precursor. The hydrothermally prepared Si-HAp fine particles have high crystallinity. However, the chemical compositional evaluation of the Si-HAp powders conducted by XPS analysis confirmed that the produced particles were Si-deficient according to the nominal Si pursued incorporating in the PO43− sites of the HAp lattice cell, and it the presence of silicon in the remaining solutions was confirmed by wet chemical analyses conducted by ICP. The lack on the Si incorporation in the hexagonal HAp structure is provoked by the formation of highly soluble intermedium species caused by the TMAS precursor, such as SiO44− [Si(OSi)3(O−)], N(CH3)4OH·5H2O and C4H13NO·3H2O. These species reduce the concentration of the solute (SiO44− ions) at the supersaturation stage reached under hydrothermal conditions, which avoids the incorporation of the nominal Si content added to the reaction system during the crystallization stage. Nevertheless, the structural analyses conducted on all the Si-HAp particles prepared revealed that reduced Si content produced the release of OH-ions simultaneously.
The gradual incorporation Si did not affect the diffraction pattern of Si-HAp prepared with TMAS, only a slightly decreased intensity was revealed due to the decrease of crystallite size. However, the diffraction peaks show a small displacement to low 2θ angles coupled with a peak broadening resulted gradually increase of the Si content incorporated into the HAp structure. Moreover, TEM observations revealed that the Si-HAp particles have a morphology that resembles rod-like shape with average particle size dimensional (length) 80 and 35nm. In particular, a significant content of Si (10mol%) incorporated in the reaction system limited the particle coarsening during the crystallization stage, it might be due to the formation of soluble silicate species that increase the molar volume of the embryos at the solute supersaturation stage, this phenomena preferentially occurred at when huge contents of Si over 10mol% were added. Additionally, the XPS and Raman analysis confirmed the uptake of Si in the HAp structure, which is remarkable for the samples 16Si-HAp and 20Si-HAp. Finally, SEM observation of the compacts sintered at 1200°C showed the formation of small glassy areas containing SiO2 between the HAp grains for the sample prepared with the highest content of 20mol% Si.
The authors acknowledge to Instituto Tecnológico Nacional de Mexico (TecNM) for supporting the present research work through the research grant 6273.17. P.Z.M.V. and J.C.R.A. are indebted to CONACYT-SNI. B.M. is grateful to the National Council for Science and Technology of Mexico, for its financial support as PhD scholarship. Many thanks are also given to Professor T. Yamamoto and Ph. D.T. Matsuzaki from the Centre for Advanced Marine Core Research, Kochi University, Japan, for their assistance with FE-scanning electron microscopy. Ph. D.T. Kono at Kochi Prefectural Industrial Technology Center, Kochi Japan, is gratefully acknowledged for his valuable support in conducting XRD characterization of the specimens.

