






Introducción: La retinosis pigmentaria es la forma hereditaria y crónica más común de distrofia retiniana. Esta condición se caracteriza inicialmente por la afectación progresiva de los fotorreceptores y, posteriormente, de otras capas de la retina. En la exploración ocular esta situación se traduce como palidez del disco óptico, disminución vascular y depósitos de pigmento en laretina.
Caso clínico: Se presenta el caso de un paciente masculino de 15 años de edad con una historia de 6 meses de evolución caracterizada por ceguera nocturna y disminución de la visión lateral temporal superior en ambos ojos.
Conclusiones: Este tipo de padecimiento ocular distrófico, genético y progresivo comienza durante la adolescencia y condiciona una discapacidad visual.
Background: Retinitis pigmentosa is the most common chronic and inherited condition of retinal dystrophy. The progressive involvement of retinal photoreceptors and other layers characterize this condition. This situation results in optic disc pallor and retinal pigment deposition vascular attenuation.
Case report: We present the case of a 15-year-old male with a history of 6 months evolution characterized by night blindness and bilateral impairment of superior temporal vision.
Conclusions: This type of dystrophy is a genetic and progressive eye condition that begins during adolescence and produces visual impairment.
Pagina nueva 1
1. Introduction
Retinitis pigmentosa (RP) is a heterogeneous group of hereditary retinal dystrophies characterized by abnormalities in the rods and, rarely, of the cones, as well as visible pigment deposits in the fundus.1 It mainly affects males from 30-50 years of age. The prevalence in the U.S. is 1/4000 persons.2 Diagnosis may be suspected due to the presence of decreased night vision, altered visual fields, and progressive loss of peripheral vision.3 It may be confused with glaucoma, papillary atrophy or other eye diseases.
Clinical presentation and severity vary based on inheritance pattern. X-linked RP generally is manifested between 0 and 6 years of age, whereas autosomal dominant RP presents a later onset. Examination of the fundus is characterized by the presence of arteriolar narrowing, intra-arterial pigmentation and loss of peripheral retinal epithelial pigmentation. With degeneration progression, there is loss of the intraretinal pigment, accumulation of melanin and appearance of bone spicules. As the disease advances, the attenuation and pallor of the optic nerve worsens and central visual acuity also may be affected in late stages due to the appearance of macular edema.4
Because of the restrictions caused by RP and the visual disability that it may cause the patient, timely identification is of vital importance so that the patient and family are aware of the genetic and progressive nature of the disorder. There is currently no treatment that will halt the ocular degenerative changes and subsequent decrease of visual acuity. During adolescence this condition may be associated with endocrine, neurological or hearing disorders, among others, which may constitute syndromes.5 The objective of this article was to present a case of an adolescent patient with RP with the purpose of raising awareness about this disease that will result in visual disability.
2. Clinical case
We present the case of a 15-year-old male without clinically significant personal or family medical antecedents. He was seen due to loss of bilateral visual acuity at night, which initiated 6 months prior, and bilateral decrease of superior temporal lateral vision 3 months prior. The patient is the product of a second gestation with a birth weight of 2950 g. On physical examination his height was 170.8 cm, weight 62 kg (BMI 21.2, between the 50th and 75th percentiles). On eye examination he had a visual acuity of 20/60 in the right eye (OD) and 20/40 in the left eye (OS). The ETDRS (Early Treatment Diabetic Retinopathy Study) method was applied and it did not correct with stenopeic glasses. Similarly, no refractive defect was detected. For refraction we used tropicamide 1% as a cycloplegic agent. Fundoscopic examination of both eyes revealed attenuation and thinning of the retinal vessels, waxy pallor of the optic disc and atrophy with hyperpigmentation in peripheral areas of the retina (Fig. 1). Goldmann perimetry showed a loss of –30.06 dB in OD and -27.38 dB in OS (Fig. 2), with negative hemifield test for glaucoma. To avoid variability of the thresholds, the test was performed by an optometrist who prevented patient fatigue and fixation errors. A fluorangiography of the fundus was carried out and revealed irregular areas of hypofluorescence in the peripheral retina with areas of hyperfluorescence in the macular region in both eyes (Fig. 3). In the electrophysiological analysis of both eyes a decrease in the electrical responses of the cones and rods ten times less than the standard deviation of the average for the patient’s age was determined. With diagnostic evidence for RP, optical coherence tomography (OCT) was done, which reported macular atrophy with loss of photoreceptors in both eyes (Fig. 4). The case analysis was completed when systemic disorders associated with RP were ruled out.
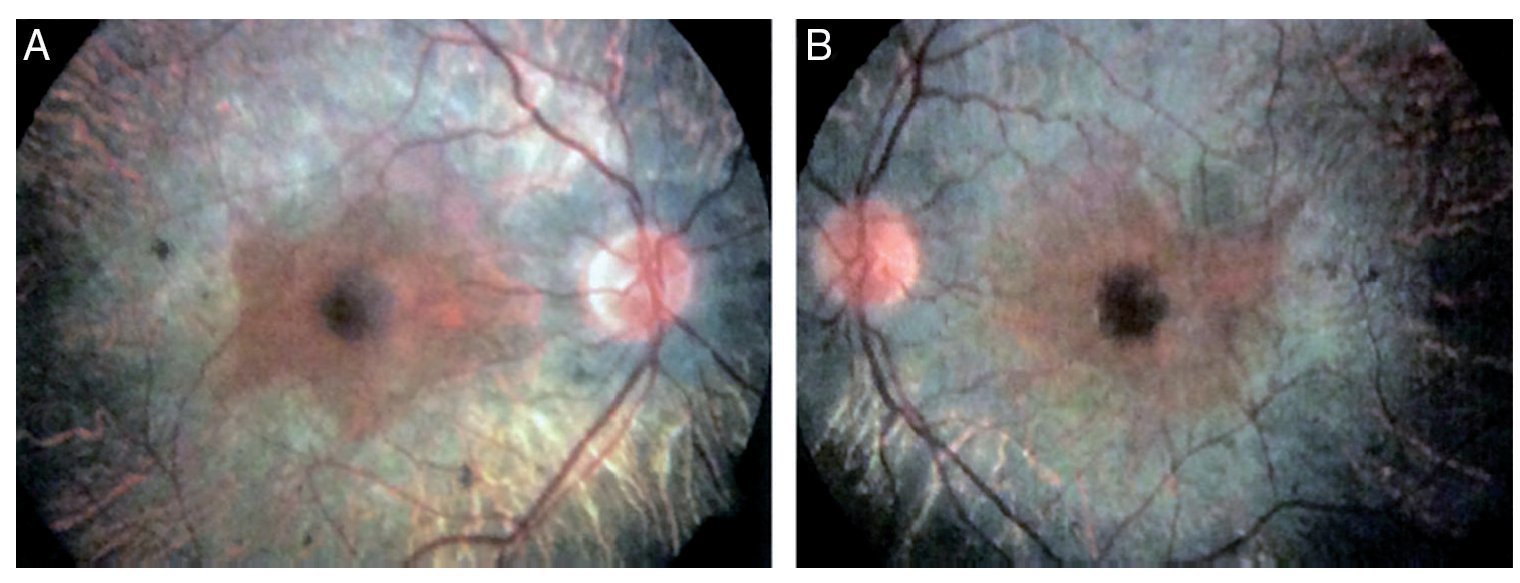
Figure 1 Fundoscopic analysis of the right (A) and left (B) eyes where abnormalities characteristics of retinitis pigmentosa are observed.
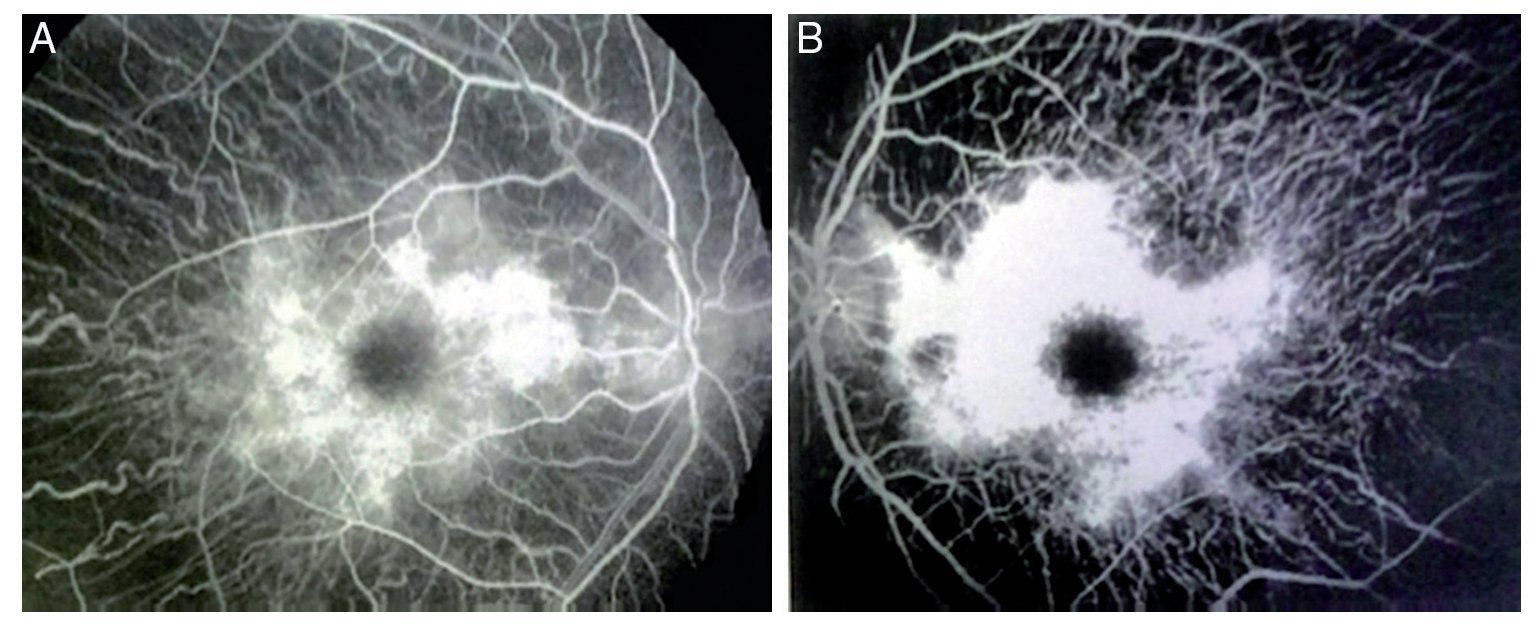
Figure 2 Goldmann campimetry for right (A) and left (B) eyes. Loss of -30.06 dB in OD and –27.38 dB in OS was determined.
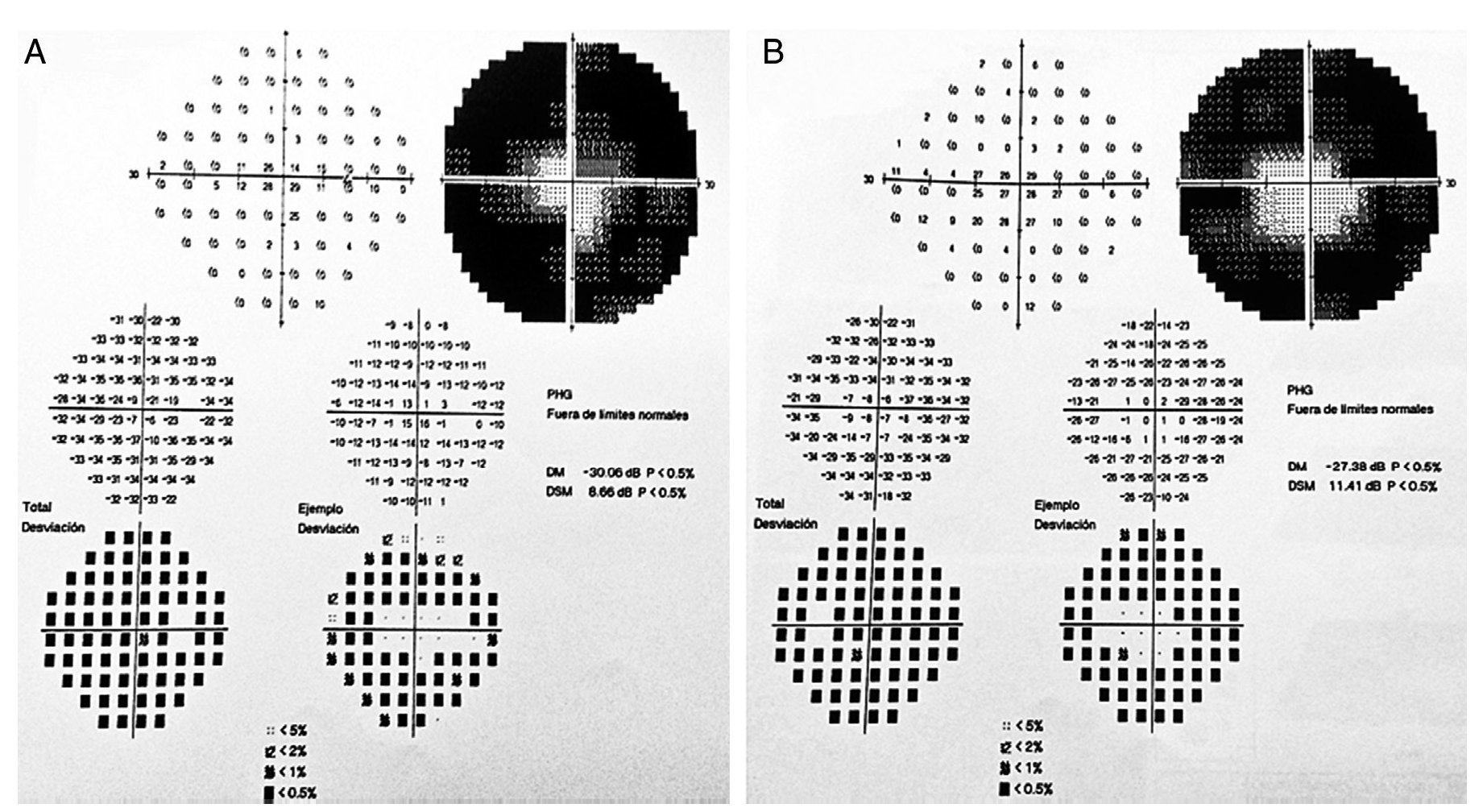
Figure 3 Fluorangiography of both eyes (A, right eye and B, left eye) showing alterations of this condition.
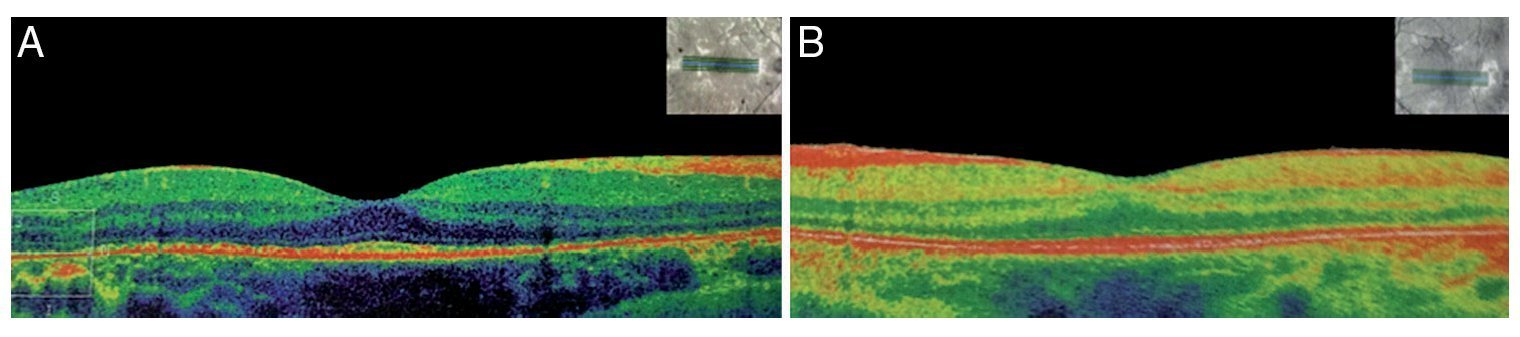
Figure 4 Optical coherence tomography (OCT) (A) right eye and (B) left eye with decrease of the photoreceptors and macular atrophy of both eyes (angle of exploration 0°, spacing 0.25 mm, length 6 mm).
3. Discussion
RP in children commonly manifests as difficulty adapting to night vision. Patients are unable to distinguish shapes and objects along with decrease in peripheral vision, photopsia and change in the perception of blue and yellow. However, there is also a common variety of syndromic forms. The most common are Usher syndrome in which deafness is associated with RP and Bardett-Biedl syndrome accompanied by obesity, polydactyly, hypogonadism and cognitive deficit.6 Differential diagnosis should be established with maculopathy in “bull’s eye”, which is characterized by blurred vision and without sharpness caused by irregular macular pigmentation, affecting 1% of the pigment epithelium and extending to both sides of the optic nerve. It is bilateral and symmetrical with an incidence of 1/10,000 persons with greater frequency in adolescents. Other conditions that cause pallor of the papilla are Leber optical neuropathy, Kjer optical atrophy, Wolfram syndrome and Behr syndrome. Chorioderemia was also ruled out. This condition is characterized by loss of the retinal epithelial pigment as well as the choriocapillary layer, although the internal retina and the optic nerve remain normal, generally indicating myopia. Currently, 45 loci/genes have been identified that cause the nonsyndromic forms of RP, i.e., autosomal dominant, autosomal recessive, X-linked and digenic.7 Mutations in the RP1, RP2 and RP3 genes associated with unilateral RP in families have been proven.8,9
In this case, clinical diagnosis was established due to the presence of decreased night vision, peripheral vision defects, lesion of the fundus, abnormally low tracings on electroretinography and progression of all symptoms. OCT was used for obtaining morphological information of the retina, supporting the diagnosis.10 Genetic mapping study is recommended to determine if there is an association with autosomal dominant, recessive or X-linked inheritance in order to provide better genetic counseling. The present case could be classified as sporadic because the patient is the first member of the family with this disorder and no linkage to inheritance was determined.
There is currently no treatment to halt the progression of the disease or to reverse the pathogenic process. The therapeutic approach is limited to delaying progression by protection from sunlight and multivitamin supplementation as well as the use of optical and electronic devices such as night vision goggles and treatment of any complications that arise.11
Ethical disclosures
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of data. The authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consent. The authors must have obtained the informed consent of the patients and /or subjects mentioned in the article. The author for correspondence must be in possession of this document.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 6 March 2015;
accepted 1 June 2015
☆ Please cite this article as: Treviño Alanís MG, Escamilla Ocañas CE, González Cerna F, García Flores JB, Moreno Treviño M, Rivera Silva G. Retinosis pigmentaria en un adolescente. Bol Med Hosp Infant Mex. 2015. http://dx.doi.org/10.1016/j.bmhimx.2015.06.001
* Corresponding author.
E-mail:gerardo.rivera@udem.edu (G. Rivera Silva).