


La retinosis pigmentaria es la forma hereditaria y crónica más común de distrofia retiniana. Esta condición se caracteriza inicialmente por la afectación progresiva de los fotorreceptores y, posteriormente, de otras capas de la retina. En la exploración ocular esta situación se traduce como palidez del disco óptico, disminución vascular y depósitos de pigmento en la retina.
Caso clínicoSe presenta el caso de un paciente masculino de 15 años de edad con una historia de 6 meses de evolución caracterizada por ceguera nocturna y disminución de la visión lateral temporal superior en ambos ojos.
ConclusionesEste tipo de padecimiento ocular distrófico, genético y progresivo comienza durante la adolescencia y condiciona una discapacidad visual.
Retinitis pigmentosa is the most common chronic and inherited condition of retinal dystrophy. The progressive involvement of retinal photoreceptors and other layers characterize this condition. This situation results in optic disc pallor and retinal pigment deposition vascular attenuation.
Case reportWe present the case of a 15-year-old male with a history of 6 months evolution characterized by night blindness and bilateral impairment of superior temporal vision.
ConclusionsThis type of dystrophy is a genetic and progressive eye condition that begins during adolescence and produces visual impairment.
La retinosis pigmentaria (RP) representa un grupo heterogéneo de distrofias retinianas hereditarias que se caracterizan por anormalidades en los bastones y, de forma más rara, en los conos, así como depósitos de pigmento en la retina visibles en el fondo del ojo.1 Afecta principalmente a hombres de entre 30 y 50 años de edad. La prevalencia en los Estados Unidos es de 1 de cada 4,000 personas.2 El diagnóstico puede sospecharse por la existencia de nictalopía, campos visuales alterados y pérdida progresiva de la visión periférica.3 Puede confundirse con glaucoma, atrofia de papila u otras enfermedades del ojo.
La presentación clínica y severidad varía con base en el patrón hereditario. La RP ligada al cromosoma X generalmente se manifiesta en edades comprendidas entre los 0-6 años, mientras que la RP con patrón autosómico dominante tiene un inicio más tardío. El examen de fondo de ojo se caracteriza por la presencia de estrechez arteriolar, pigmentación intraarterial y pérdida del pigmento epitelial retiniano periférico. Con la progresión de la degeneración hay pérdida del pigmento intrarretiniano, acumulación de melanina y aparición de espículas óseas. Conforme avanza la enfermedad, la atenuación y palidez del nervio óptico se agrava, y la agudeza visual central también puede afectarse en etapas tardías debido a la aparición de edema macular.4
En virtud de las restricciones que la RP produce y la discapacidad visual que puede ocasionarle al paciente, es de vital importancia identificar este padecimiento a tiempo con la finalidad de concientizar tanto al paciente como a sus familiares de la naturaleza genética y progresiva de las alteraciones, ya que no existe hasta el momento un tratamiento que detenga los cambios degenerativos oculares y la consecuente disminución de la agudeza visual. Durante la adolescencia esta condición puede asociarse con padecimientos endocrinos, trastornos neurológicos, auditivos, entre otros, que podrían constituir síndromes.5 El objetivo de este artículo fue presentar un caso de un paciente adolescente con RP, con el propósito de concientizar sobre esta enfermedad que terminará en una minusvalía visual.
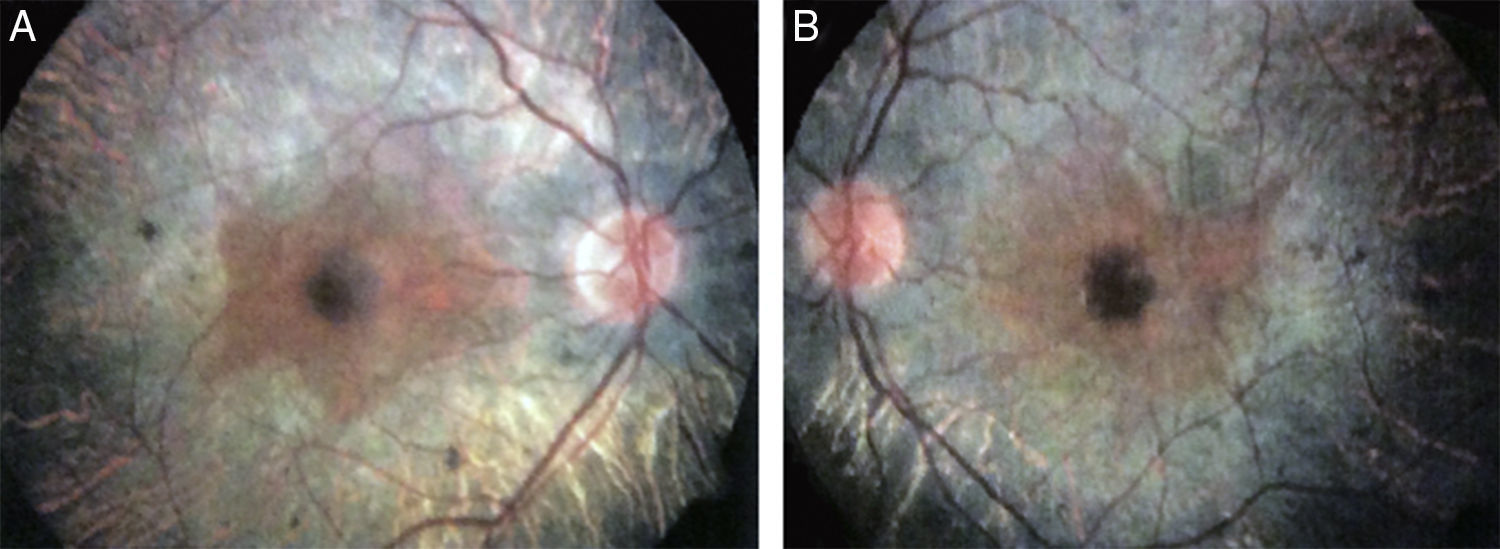
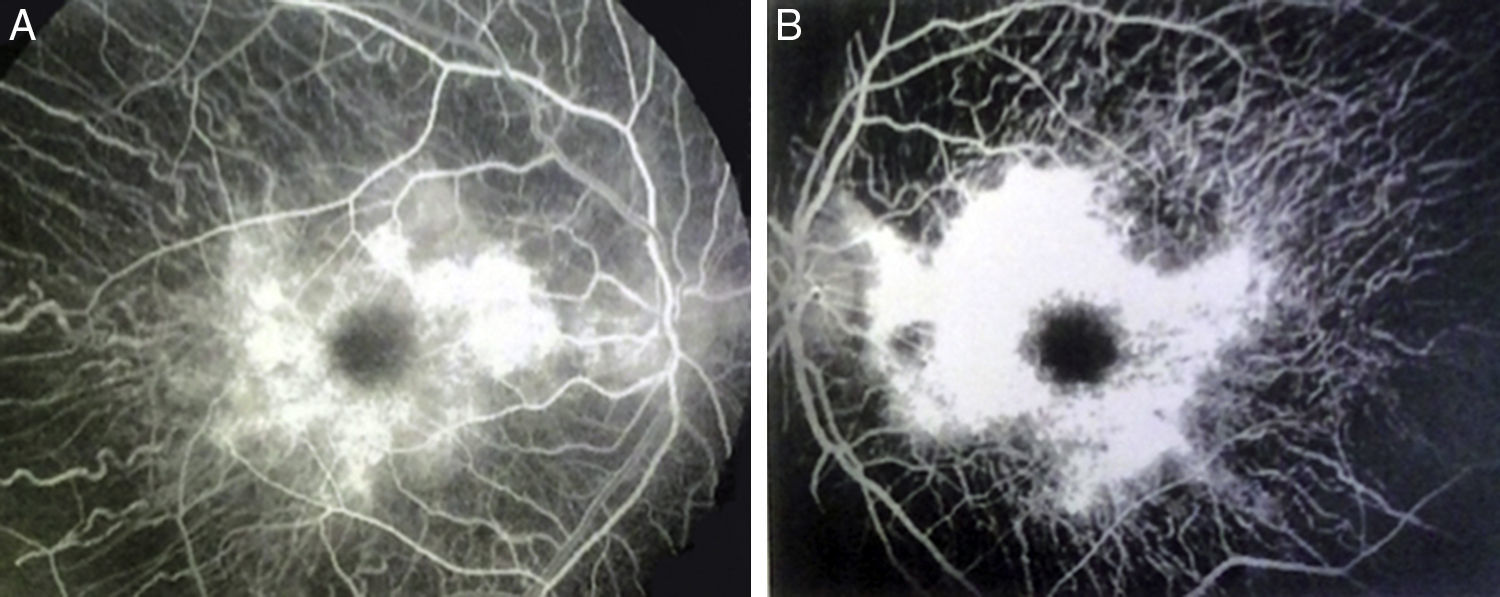
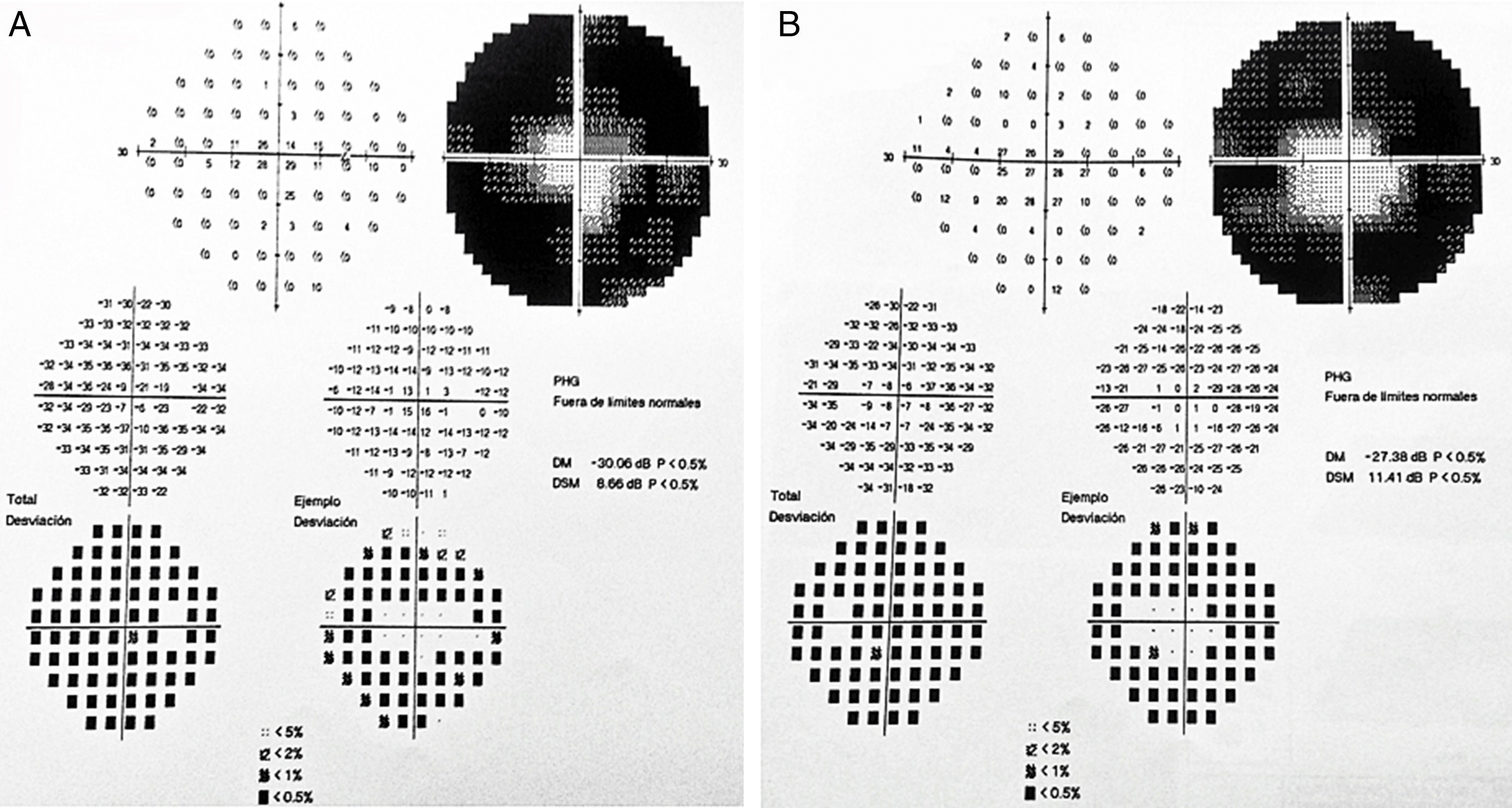
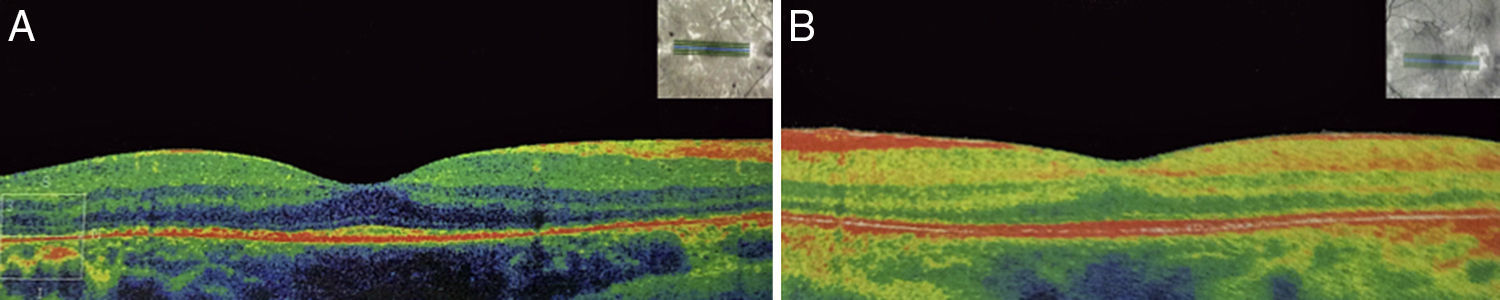
2Caso clínicoAdolescente masculino de 15 años de edad, sin antecedentes personales patológicos ni familiares de interés en la historia familiar. Acudió a consulta por la pérdida de la agudeza visual bilateral nocturna que comenzó 6 meses antes, y disminución bilateral de la visión lateral temporal superior desde 3 meses antes. Producto de la gesta II, embarazo II y con un peso al nacer de 2,950g. En la exploración física se encontró talla de 170.8cm, peso de 62kg (IMC 21.2, centilo entre 50 y 75). En el examen oftalmológico presentó agudeza visual de 20/60 en ojo derecho (OD) y 20/40 en ojo izquierdo (OS). Se aplicó el método ETDRS (Early Treatment Diabetic Retinopathy Study) y no corrigió con estenopeico. Asimismo, no se detectó ningún defecto refractivo. Para la realización de la refracción se utilizó tropicamida al 1% como cicloplégico. El análisis fundoscópico de ambos ojos reveló atenuación y adelgazamiento de los vasos retinianos, palidez cérea del disco óptico y atrofia con hiperpigmentación en áreas periféricas de la retina (Fig. 1). En la campimetría de Goldmann se encontró con pérdida de -30.06dB en OD y -27.38dB en OS (Fig. 2), con resultado de la prueba de hemicampo para glaucoma negativo. Para evitar la variabilidad de los umbrales, la prueba fue realizada por un optometrista, quién evitó la fatiga del paciente y errores de fijación. Se realizó una fluorangiografía de fondo de ojo, la cual reveló zonas de hipofluorescencia irregular en retina periférica con áreas de hiperfluorescencia en región macular tanto de OD como de OS (Fig. 3). En el análisis electrofisiológico de ambos ojos se determinó una disminución de las respuestas eléctricas de conos y bastones diez veces menor de la desviación estándar del promedio para su edad. Ante la evidencia diagnóstica de RP, se realizó una tomografía de coherencia óptica (TCO) reportando atrofia macular con pérdida de fotorreceptores en ambos ojos (Fig. 4). El análisis del caso se terminó al descartarse alteraciones sistémicas asociadas a la RP.
 e izquierdo (B), donde se observan las anomalías características de la retinosis pigmentaria.")
 e izquierdo (B). Se determinó pérdida de -30.06dB en OD y -27.38dB en OS.")
 que muestra las alteraciones de esta condición.")
 y de ojo izquierdo (B) con disminución de los fotorreceptores y atrofia macular de ambos ojos (ángulo de exploración 0°, espaciamiento 0.25mm, longitud 6mm).")
La RP en niños se manifiesta comúnmente como dificultad para adaptarse a la visión nocturna, no pueden distinguir formas y objetos, disminución de la visión periférica, fotopsias y alteración en la percepción del azul y amarillo. Sin embargo, existe también una variedad común de formas sindrómicas. Las más frecuentes son el síndrome de Usher, en el que hay sordera asociada a la RP, y el síndrome de Bardet-Biedl, que se acompaña de obesidad, polidactilia, hipogonadismo y déficit cognitivo.6 El diagnóstico diferencial debe establecerse con la maculopatía en “ojo de buey”, que se caracteriza por una visión desenfocada y sin nitidez provocada por una pigmentación macular irregular que afecta el 1% del epitelio pigmentario y que llega a ambos lados del nervio óptico. Es bilateral y simétrica, y suele presentarse en 1/10,000 personas, con mayor frecuencia en adolescentes. Otras condiciones que producen palidez de la papila son la neuropatía óptica de Leber, la atrofia óptica de Kjer, el síndrome de Wolfram y el síndrome de Behr. También se descartó coroideremia. Esta condición se caracteriza por la pérdida tanto del pigmento epitelial retiniano como de la capa coriocapilar, aunque la retina interna y el nervio óptico permanecen normales; por lo general se trata de pacientes con miopía. Actualmente se han identificado 45 locus/genes causales para las formas de RP no sindrómicas, es decir, para las formas autosómica dominante, autosómica recesiva, ligada al X y digénica.7 Se han comprobado mutaciones en los genes RP1, RP2 y RP3 asociadas con una RP de presentación unilateral en familias.8,9
En este caso, el diagnóstico clínico se fundamentó en la presencia de nictalopía, los defectos de la visión periférica, las lesiones del fondo ocular, los trazos anormalmente bajos en la electrorretinografía y la progresión de todos estos síntomas. La TCO se utilizó para la obtención de información morfológica de la retina aportando complementación diagnóstica.10 El estudio con mapeo genético es recomendable para determinar si existe una asociación con la herencia autosómica dominante o recesiva o ligada al cromosoma X, y de esta manera poder brindar un mejor consejo genético. El presente caso puede ser catalogado como esporádico, ya que es el primer miembro de la familia que tiene este padecimiento y no se pudo determinar ninguna vinculación a la herencia.
Actualmente no existe tratamiento que detenga la evolución de la enfermedad o logre revertir el proceso patogénico. El abordaje terapéutico se limita a retrasar la progresión mediante la protección de la luz solar y el suplemento con multivitamínicos, además de dispositivos ópticos y electrónicos, como las gafas de visión nocturna y el tratamiento de las complicaciones que se presenten.11
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.