










Pagina nueva 1
1. Clinical history (A-13-01)
We present the case of a newborn male referred for cystic adenomatoid disease.
1.1. Family history
The patient’s mother is a 21-year-old college student, GI, CI, P0, A0 and reported one sexual partner. The 21-year-old father is an employed high-school graduate who reported a smoking habit of five cigarettes per day.
1.2. Social history
The family lives in a rented house with basic services in an urban area. Five people live in three rooms with a pet dog. The patient was reported fasting since birth. No immunizations were recorded.
1.3. Perinatal and medical history
The patient was the product of a GI with prenatal care from the second month. The mother was administered folic acid, iron and vitamins. She reported a urinary tract infection during the first trimester and cervicovaginitis in the third trimester without specifying treatment. Obstetric ultrasound at week 20 identified cystic adenomatoid disease. Fetal surgery with diagnostic fetal bronchoscopy was carried out. Bronchial atresia was ruled out and a thoraco-amniotic shunt was placed for cyst drainage. Due to fetal heart failure, laser ablation of the tumor blood flow was done to decrease its size. Three doses of cefotaxime were given to the mother as prophylaxis.
1.4. Current illness
The patient was admitted to this institution with a diagnosis of fetal cystic adenomatoid disease. The patient was born via cesarean section at 40 weeks of gestation (WG) and was intubated. Due to heart rate <60, a cycle of chest compressions was given and the patient recovered normal heart rate. Apgar was 3/7. Umbilical arterial and venous accesses were placed.
PHYSICAL EXAMINATION. Physical examination revealed a newborn male with good color.
HEAD AND NECK. Head and neck examination revealed normocephalic presentation, anterior fontanel 2 × 2 cm, and posterior puntiform. Pupils were isochoric, ear lobes were well placed, and nares were permeable. A 3.5-mm endotracheal tube was placed. Neck was cylindrical without adenomegaly. Conventional mechanical ventilation was administered.
CHEST. Chest was symmetrical. Precordium had rhythmic heart sounds without added sounds. Lung fields showed generalized hypoventilation and 2-cm left parascapular fistula.
ABDOMEN. Abdomen was soft and depressible without visceromegaly. Male genitalia were without alterations. extremities. Extremeties were intact and pulse was symmetrical.
TRANSFONTANEL AND RENAL USG. These were without signs of focal lesions or bleeding and with well-defined and symmetrical choroid plexus. Nuclei of the base and posterior fossa were normal. Kidneys were normal.
ECHOCARDIOGRAM. There was levocardia, normal venous return, atrioventricular connection and concordant atrial ventricle. Pulmonary pressure was 95 mmHg. There was severe tricuspid insufficiency, dilatation of the left cavities, and external compression of the left ventricle. Ventricular function was adequate. No shunt was seen through the foramen ovale and the permeable ductus arteriosus.
TREATMENT. Left superior pulmonary lobectomy was carried out. The patient was managed with dopamine, adrenaline and norepinephrine. High frequency ventilation and inhaled nitrous oxide at 20 ppm were delivered. Antimicrobial treatment included cefepime and amikacin. Right thoracocentesis was done. Despite interventional management, the patient presented acidosis and cardiopulmonary arrest without response to resuscitation maneuvers.
2. Case presentation
2.1. Medical Department (Dr. Jaime Nieto-Zermeño)
We present a sui géneris case highlighted by the support on the part of the Fetal Surgery Department and which constituted a complete challenge.
2.2. Fetal medicine (Dr. Rogelio Cruz Martínez)
A 21-year-old patient at 25 WG gave birth to a single male child. The mother was referred by her gynecologist due to detection of fetal hydrops and a left-side lung mass. Obstetric ultrasound noted a hyperechogenic mass with a large solid component and cystic images >1 cm in diameter, occupying almost the entire left side of the chest and deviating the mediastinum and heart towards the right side of the fetus. The lungs were noted to be completely collapsed with a lung area equaling 15% with respect to the expected area for gestational weight as well as a small amount of fluid in the lungs indicative of a pleural effusion. In the sagittal cut a large amount of ascitic fluid was observed with an intact diaphragm, large hyperechoic mass with a solid and cystic component, pleural effusion and subcutaneous edema in the chest. Therefore, the fetus had heart failure due to a large unilateral mass deviating the mediastinum to the right, severe pulmonary hypoplasia and fetal hydrops.
The possibilities of intrauterine death associated with hydrops are 100%. If a cesarean delivery was carried out, risk of neonatal death was excessively high because it was a 720-g fetus, 25 WG, with severe pulmonary hypoplasia and heart failure. The mother was offered the following options: 1) no further procedures to be carried out with the certainty that there would be intrauterine death; 2) perform a cesarean section with the risk of neonatal death >90%; or 3) carry out fetal surgery with 50% chance of success. The family accepted the third option, which had the greatest possibilities of success. It was proposed to first do a diagnostic bronchoscopy to rule out that there was communication with the tracheobronchial tree and to confirm that it was a congenital cystic adenomatoid malformation (CCAM). During the same procedure, with minimally invasive surgery, local anesthesia was then administered to the mother and a needle was placed that traversed the uterus, membranes and the central and interior part of these cysts. A shunt catheter was placed within the cysts and the other end in the amniotic cavity. This was done so that these cysts, which were connected to each other inside, could be emptied, thereby decreasing tumor size and the associated cardiac compression. The intervention was technically successful. There was no complication or rupture. The heart was seen to be less compressed, but the large solid cystic mass and significant pulmonary hypoplasia persisted. Heart failure began to decrease and improved; however, pulmonary hypoplasia persisted due to the tumor mass. This CCAM had blood flow from the pulmonary artery. A second intervention was proposed to eliminate this blood circulation and decrease the solid component of the CCAM in order to increase the size of the right lung. The intervention was done 1 week after the first, at 26 WG. Once again, it was a minimally invasive percutaneous surgery with local and intramuscular fetal anesthesia, with fentanyl, vercuronium and atropine so that the fetus would not move and cause him no pain. Laser ablation of the entire blood circulation of the mass was done. The intervention was successful in the sense that there were no significant complications. Cardiac failure was reversed completely and at 30 WG there was no pleural effusion or ascites. The right lung began to improve. Lung size increased and the tumor mass began to decrease. At 32 WG the solid component of the mass began to decrease, but the cystic component increased. What may have happened was that the catheter still in place became obstructed and the amount of fluid inside the tumor mass increased, but the solid component had decreased. Intratumoral laser resulted in improvement of the right-side tumor and there was no cardiac failure. For this reason, carrying out a third fetal intervention was considered risky. The 39-WG ultrasound demonstrated ascitic fluid. At the level of the chest there was an improved lung size. The cystic component of the mass began to increase in size, but the size of the right lung was up to 90% vs. the expected size for gestational age. Finally, the possibilities of life at 39 WG were much better with regard to what was evident at 25 WG. The fetal management situation followed with the delivery of the infant and in the following section we describe the radiological findings.
2.3. Imaging (Dra. Bertha Romero-Baizabal)
The first x-ray (January 24, 2013) was the most informative. The left lung was observed to have many large, well-circumscribed radiolucent images occupying practically the entire left lung. Mediastinal structures as well as the left lung itself were displaced towards the right side (Fig. 1). The diaphragm was apparently intact with little distribution of intestinal gas. The tracheal cannula was in good position, but lateralized towards the right. In another sequence (February 3), a left-side radiolucent image was seen in relation to a persistent pneumothorax, although of smaller dimensions. In the right lung there were changes in density seen in relation with a reticular type pattern, which did not rule out the possibility of an infectious process. Therefore, the radiological diagnoses were type 1 congenital lung malformation of the airway, bilateral pulmonary pneumonic process and left pneumothorax. The last films were taken after resection of the upper lobe of the lung. There was a very small left lung, which allowed re-expansion of the right lung. There was a residual pneumothorax, which is common for these patients (Fig. 2).
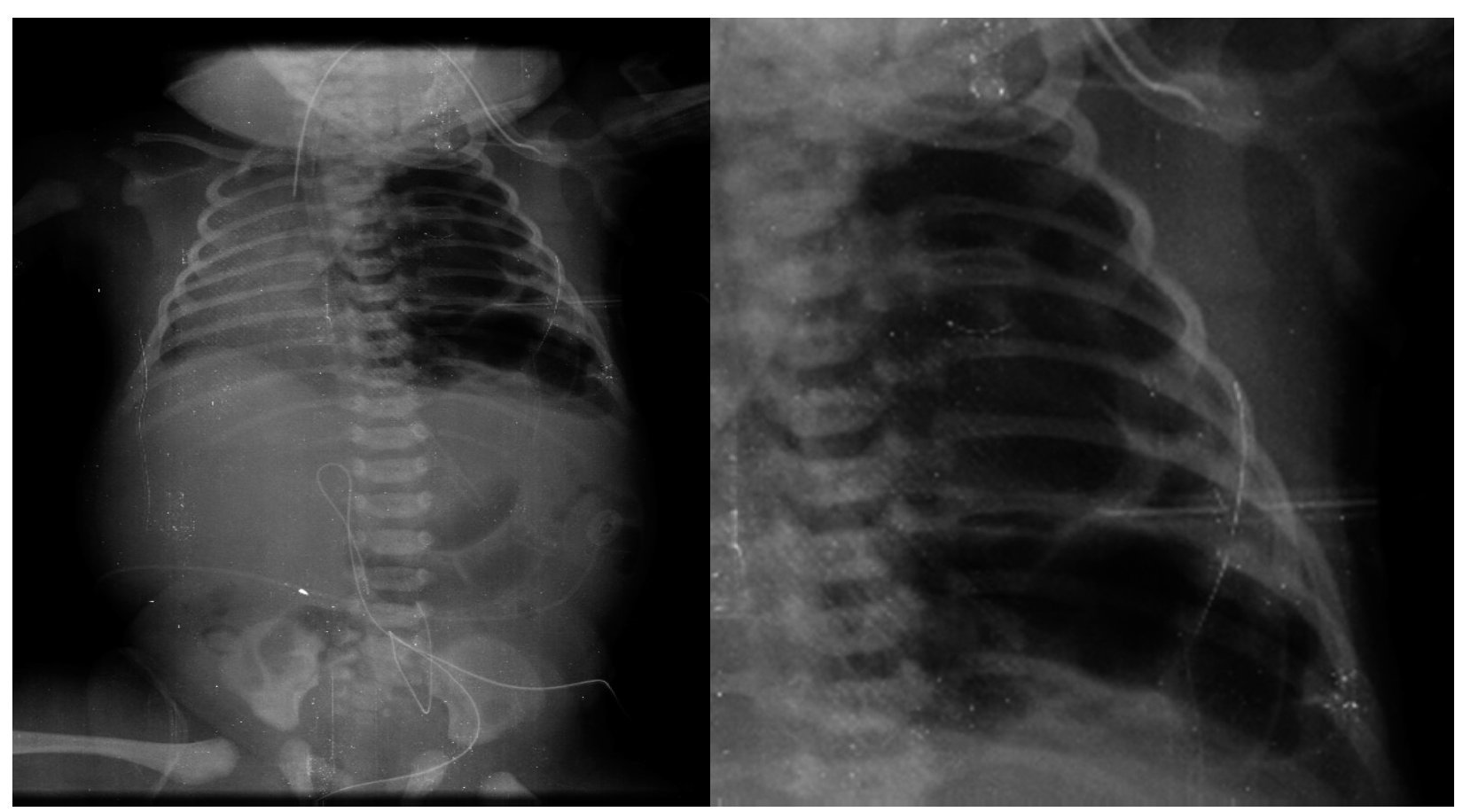
Figure 1 Cystic images covering the entire left lung and mediastinal structures shifted to the right.
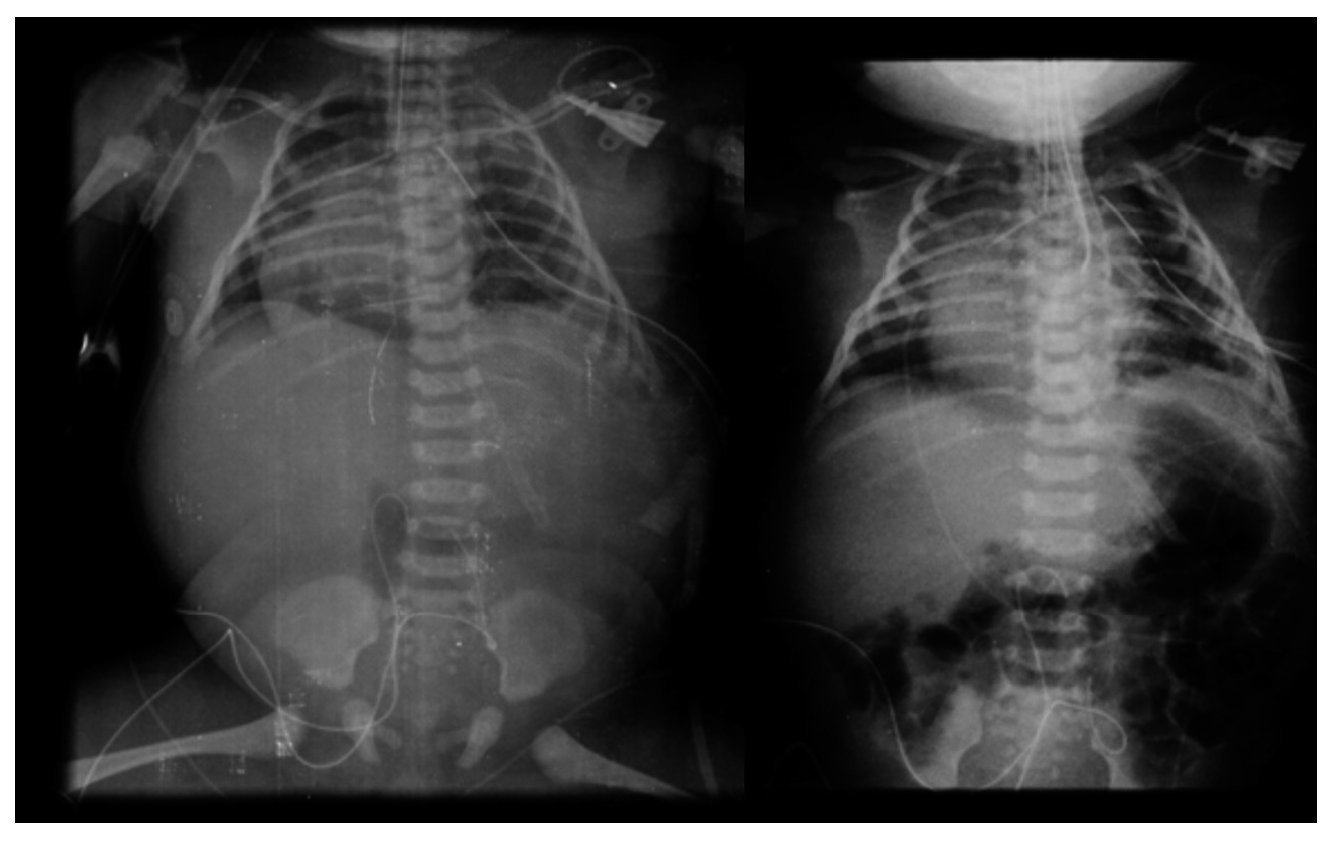
Figure 2 Postoperative study where cardiomegaly with right shift is shown. There is also persistence of some images of cystic aspect in the left lung.
3. Discussion
3.1. Pediatric surgery
(Dra. Ebenezer Viridiana Cruz Romero)
We discuss the case of a patient with CCAM with a history of heart failure and fetal hydrops for whom novel management and treatment for a common malformation are administered. This patient presented the following syndromatic diagnoses:
• Respiratory distress syndrome with increase in respiratory rate, generalized hypoventilation, hypoxemia, hypercapnia and respiratory acidosis
• Air leak syndrome characterized by hypoventilation, tympanism, and radiographic imaging showing a pneumothorax
• Systemic inflammatory response syndrome characterized by persistent tachycardia, fever of 38 °C and leukopenia
• Condensation syndrome according to fine crackles in the right hemithorax, dullness to percussion and absence of left base sounds
• Acute renal insufficiency due to increase in reatinine up to 3.3 mg/dl
• Persistent metabolic acidosis
On this basis, the nosological diagnoses are mentioned as follows:
1) Cystic adenomatoid malformation whose growth caused fetal heart compression with non-immune hydrops, heart failure and recurrent pneumothorax
2) Pulmonary artery hypertension with pulmonary pressure quantified up to 95 mmHg, which causes persistent hypoxia and did not respond to the administration of inotropic amines, pulmonary vasodilators and nitric acid
3) Health care-associated infection with ventilator-associated pneumonia and intrauterine manipulation of the airway along with orotracheal intubation from birth, complicated by septic shock due to systemic inflammatory response requiring aminergic support without adequate response
4) Multiple organ failure due to respiratory, cardiovascular, renal and hematological failure
It is important in this case to stress three principal points: prenatal management, medical management and surgical treatment. Prenatal management was carried out at 25 WG. This is very important because prenatal diagnosis of these types of malformations is only done in 20-30% of pregnancies. In this case it allowed for follow-up to be performed, identifying the poor prognostic factors with a fetal hydrops and heart failure in a fetus <30 WG. This allowed for the decision to be made to carry out fetal intervention. The modern perinatal team includes specialists in maternal/fetal health and experts in diagnostic imaging by ultrasound or magnetic resonance.1,2 In addition, it is important for an echocardiogram to be performed by a subspecialist in fetal echocardiography. Surgical procedures can be performed by a maternal/fetal physician or pediatric surgeon trained in fetal surgery. These cases should be discussed together with the neonatologists who will be present at birth and the possible specialists involved in the management so as to plan the site of birth, the procedures that will be carried out during resuscitation and the neonatal intensive care units (NICU) and the ideal time for performing the definitive surgical procedure. In this case, thoraco-amniotic bypass was done first. The largest case series reported with this treatment for CCAM is only 11 patients. This procedure has been shown to increase patient survival from 33-75%. In all reported series, the poorest prognostic factor has been the presence of fetal hydrops.3 After bypass, the principal parameter for evaluating fetal improvement is measurement of the ratio of the lung-cephalic perimeter. Laser ablation was then carried out because the fetus had heart failure. A literature review reported only 16 patients with pulmonary malformation were found who were treated with laser ablation: nine had lung sequestration and only seven had CCAM, reporting a survival rate of 28.6%. There are currently no prenatal therapy guidelines for this type of fetal lesion. Different alternatives have been proposed such as open fetal surgery, steroid administration by intrauterine puncture, alcohol embolization, simple aspiration of the cysts, and even the administration of OK432. In Mexico, Hospital Infantil de México Federico Gómez (HIMFG) has cared for the first infants with a history of fetal intervention and the latter is the only case with CCAM.
Regarding medical treatment, the patient was born at term, with respiratory difficulty being present from birth. He required orotracheal intubation and high ventilatory parameters. Pulmonary pressure was 95 mmHg with persistent hypoxemia, for which it is believed that added to the high frequency oscillatory ventilation, use of nitric oxide and sildenafil were necessary from the beginning as well as other therapeutic alternatives such as low-dose vasopressin. Currently, the institution does not have extracorporeal membrane oxygenation, which would have been a useful tool. Also, as has been mentioned, the patient presented with a potential infection due to intrauterine manipulation and with intensive care requirements. Aminergic management was dynamic because the challenge was to maintain an adequate systemic arterial pressure and to decrease pulmonary pressure. This management was guided by continuous monitoring with pulse oximetry, arterial and venous blood gases and additional cardiac ultrasound.4-6
The primary objective in the immediate neonatal period is drainage of fluid or air collection and ventilatory support to correct the hemodynamic status. Placement of the chest tube and necessary maneuvers to correct functionality was considered adequate because the patient had poor response to conservative treatment. Resection of the lesion was done with the anticipation of clinical improvement due to the decrease of the compression of the left ventricle with transient decrease in the pulmonary pressure of up to 30 mmHg.
Based on the above discussion, the final clinical diagnoses were as follows:
• Term newborn with weight appropriate for gestational age
• History of non-immune fetal hydrops and heart failure
• Type 1 cystic adenomatoid malformation
• Severe pulmonary hypertension
• Nosocomial pneumonia
• Septic shock refractory to amines
• Multiorgan failure
4. Pathology
(Dra. María Argelia Escobar Sánchez)
A surgical specimen corresponding to the upper lobe of the lung is observed. The specimen weighed 80 g and had opaque, dull, and congestive pleura. On serial cuts there were numerous cysts seen with an average diameter between 2 and 5 cm. Between the cysts there were areas of pulmonary parenchyma with serious congestion. There is a larger cyst with a diameter of 6 cm and whose internal surface is shiny and trabeculated (Fig. 3). Other smaller cysts were separated by septum of connective tissue, which was histologically corroborated. The malformation was constituted by numerous cysts covered by columnar epithelium and ciliated cylindrical epithelium, which alternated with some mucosal cells. These mucous-producing cells were reminiscent of intestinal glands. Between these, septae of fibroconnective tissue with some compressed cells were observed. Between the cysts covered by this epithelium we also observe the presence of smooth muscle (Fig. 4).
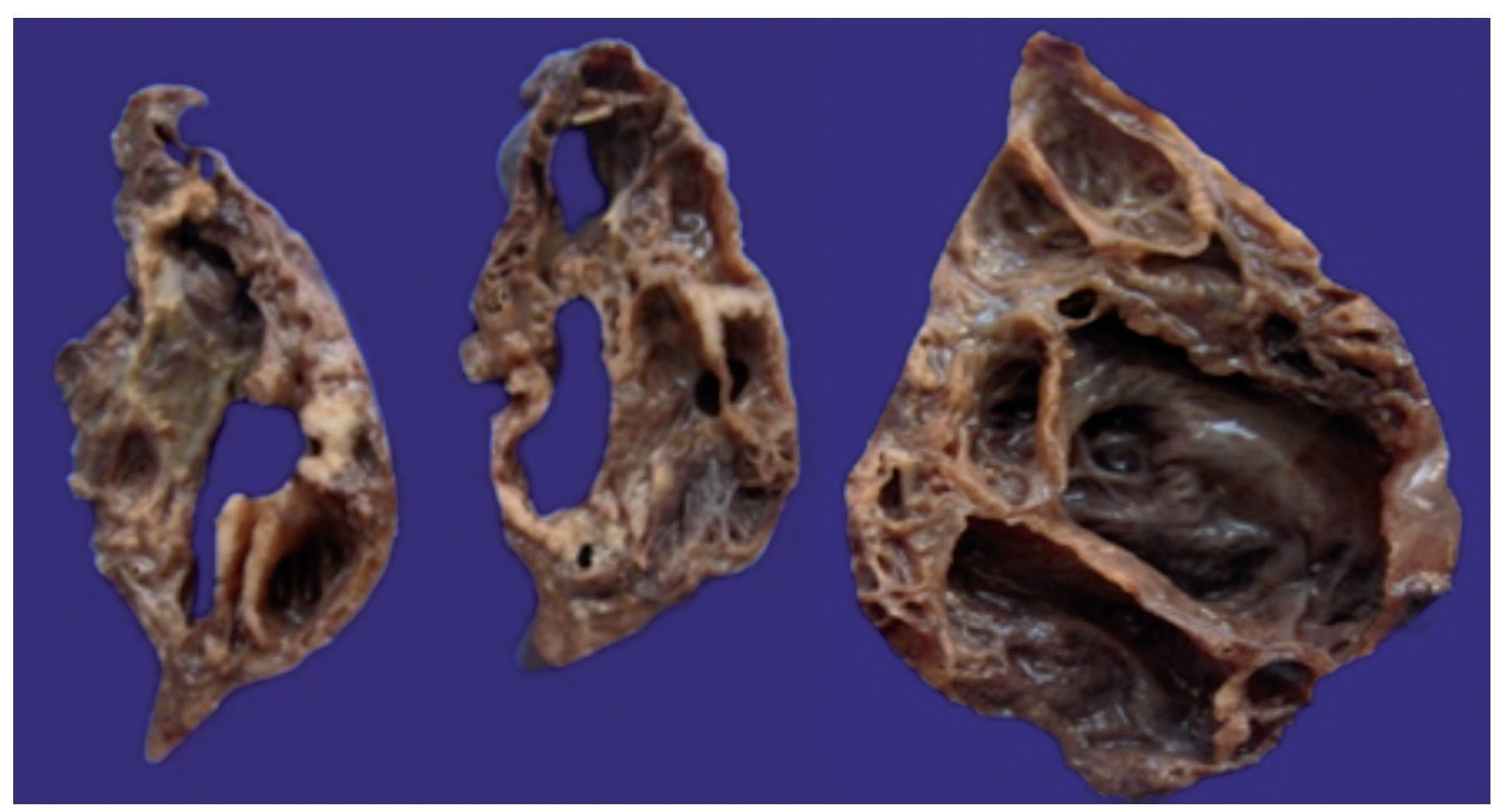
Figure 3 Macroscopic photograph of the left upper pulmonary lobe showing numerous cysts (the largest 6 cm in diameter).
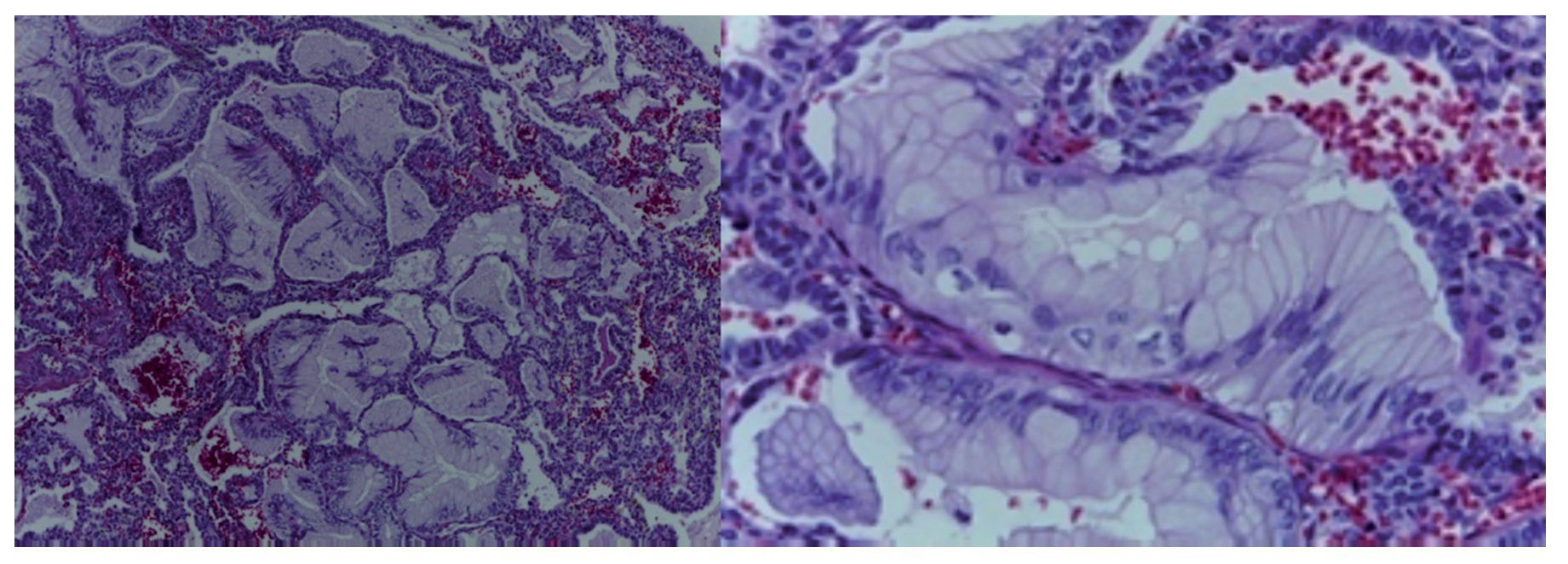
Figure 4 Cysts lined with mucoproducing columnar cells and presence of smooth muscle (HE 40×).
In addition to the result of the left upper lobectomy, the placenta was also received with a weight of 700 g, measuring 21 × 20 cm. The membranes were intact. On the maternal face the cotyledons were observed without changes and on the fetal face the membranes were congestive with central insertion of the cord. The placenta was monoamniotic and monochorionic (Fig. 5). Different cuts of the placenta showed the cord with blood vessels without alterations, intact congestive membranes and the placenta with villi representative of the third trimester of pregnancy with the presence of hyalinized thrombi and calcifications representative of gestational age (Fig. 6).
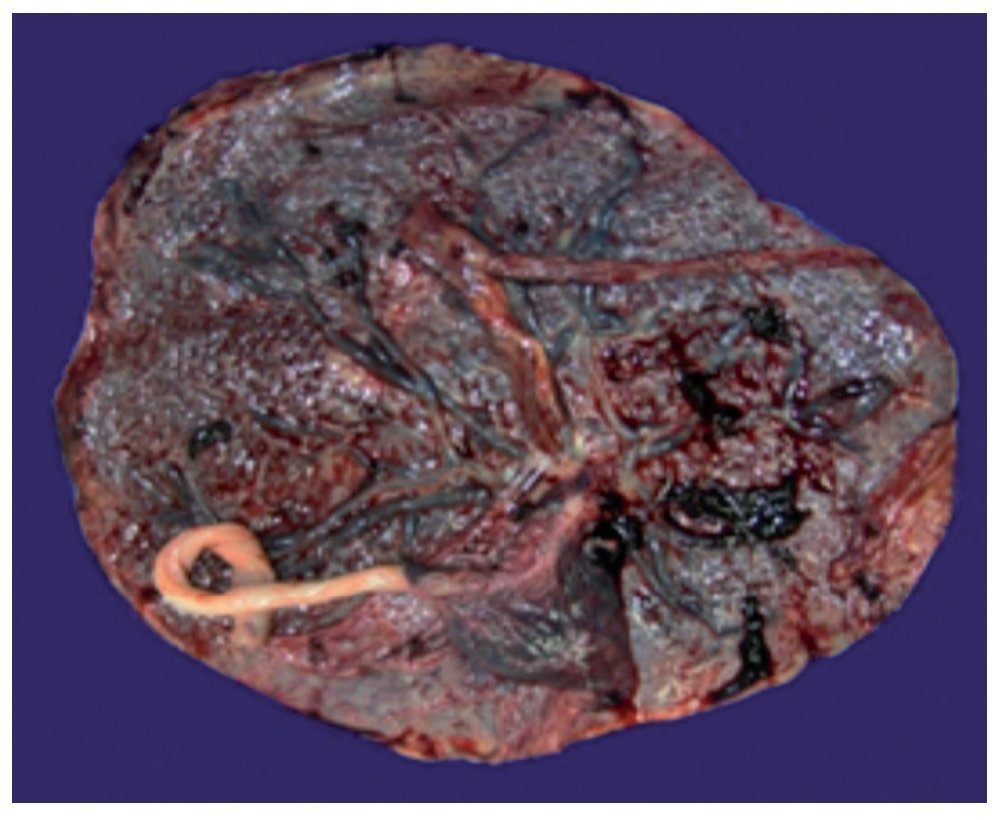
Figure 5 Macroscopic photograph of the placenta.
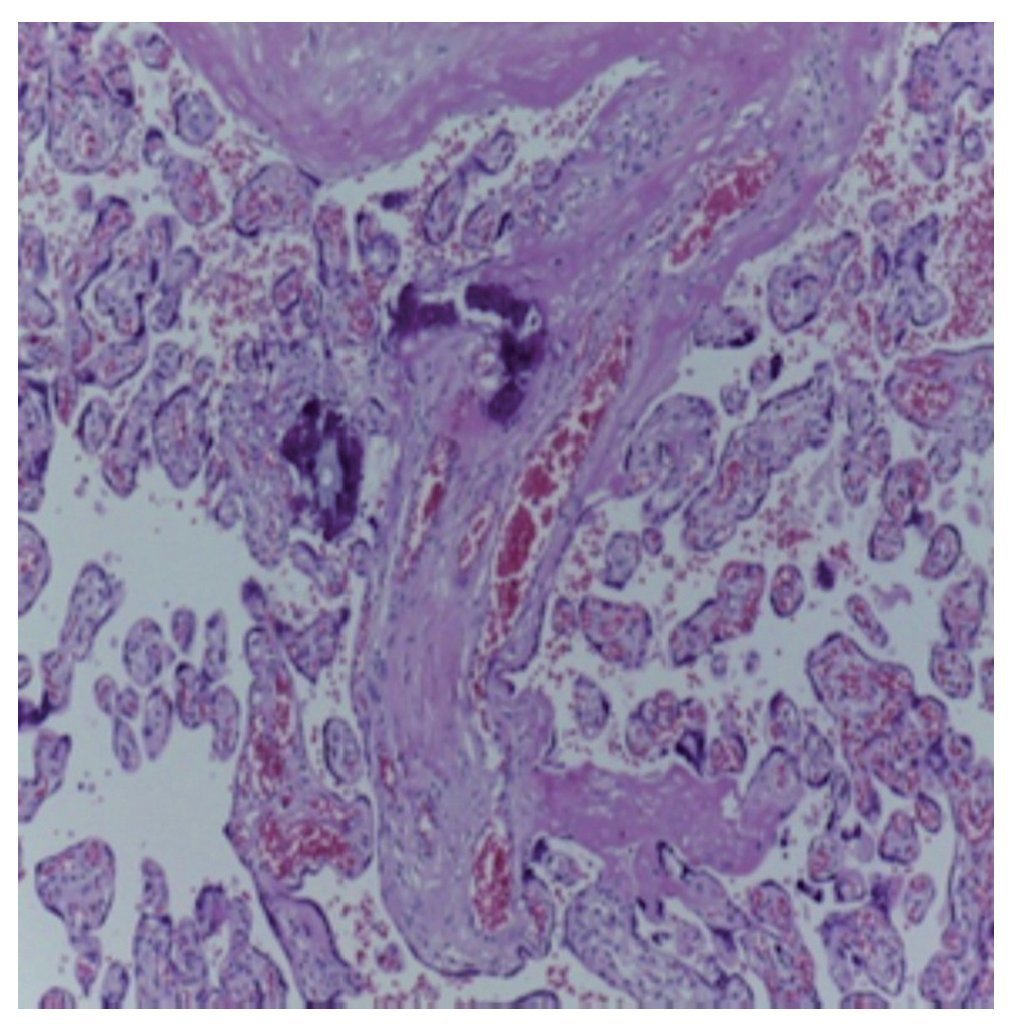
Figure 6 Microscopic photograph of the placenta.
With these findings, diagnosis of type 1 congenital pulmonary malformation of the airway was made or what has previously been referred to as cystic adenomatoid disease and placenta of the third trimester with calcifications and without alterations. The exterior habitus showed a male product weighing 3500 g vs. an expected weight of 3100 g. There were extensive areas of desquamation and a sutured wound at the level of the chest as well as two orifices that corresponded to pleural tubes localized in the fifth and tenth left intercostal space. At the time of evisceration there was no presence of pneumothorax.
In the specimen that corresponded to the cardiopulmonary block, the heart was seen with the large vessels of normal characteristics, the pulmonary area is anterior, the aorta is posterior, and the pulmonary venous circulation is normal. An important dilatation of the right ventricle is noteworthy. The right lung showed its three lobes without alteration and only the left residual lung after the surgical event was seen. On higher magnification, it was observed that the pleural surface was irregular, congestive and with normal sutures and without hemorrhage, i.e., there were no alterations after surgery. In the cut corresponding to the diaphragm, significant pulmonary congestion was observed as well as inflammatory infiltrate constituted by lymphocytes and multinucleated foreign body giant cells. The pleura also showed thickening, lymphocytic inflammatory infiltrate and numerous multinucleated giant cells that extended to the residual pulmonary parenchyma where it was observed that the alveoli were collapsed and there was the presence of granuloma secondary to the surgical event. The right lung showed intra-alveolar inflammatory infiltrate constituted by neutrophils and some areas of necrosis, i.e., this patient also had an infectious process of the right lung. There was collapse of some alveoli and in other areas there was extensive pulmonary hemorrhage as well as intra-alveolar edema and the presence of vascular pulmonary hypertension localized in the arteries and arterioles of small caliber <200 µm. In these, increased connective tissue in the adventitia could be seen, which was evident with special Masson stains and elastic fibers (Fig. 7) as hypertrophy of the muscle and endothelial cell hyperplasia. The residual pulmonary parenchyma was hypoplastic, i.e., less than six alveoli, counting from the pleura to the terminal bronchus. These presented compression, for which there was interstitial cellular proliferation. The heart weighed 50 g vs. an expected weight of 20 g and there was significant hypertrophy and dilatation of the right cavities. On opening of the cavities a foramen ovale of 0.2 cm in diameter was found. Histologically, only some hypertrophic nuclei of the cardiomyocytes were seen, and the rest were without alteration. The liver was increased in size with a weight of 200 g vs. an expected of 127 g; macroscopically it showed areas of congestion. Histologically, significant congestion of the sinusoids located in the lobules and around the central veins was observed. On higher magnification, extravasation of the erythrocytes was identified, i.e., venous flow obstruction.
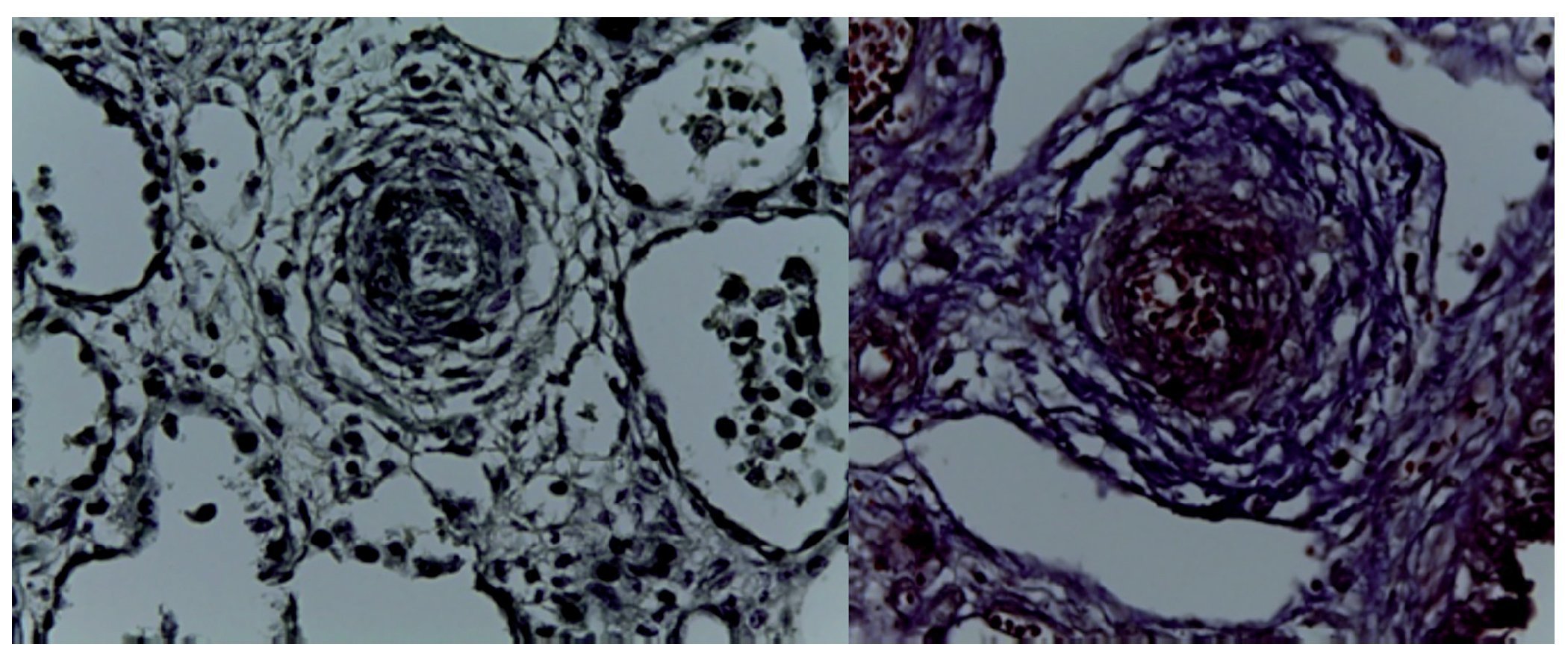
Figure 7 Elastic fibers and Masson (40×).
With these findings, a type 1 congenital malformation of the airway was diagnosed according to the size of the cysts and the presence of smooth muscle located between the cysts. It should be mentioned that this malformation is most commonly seen in ~70% of the cases. Three types were previously described. It is currently known that there are four types. Stocker described these in 2002 and they are classified from 0 to 4 where 0 is incompatible with life and 4 is the extreme.
The patient presented a left superior postlobectomy status, acute and chronic pleuritis, foreign body granulomatous reaction, right basal pneumonia and Rabinovitch grade B pulmonary vascular disease. There was also cardiomegaly, permeable foramen ovale with persistence of the permeable ductus arteriosus of 0.3 cm, congestive heart failure, congestive hepatomegaly and venous flow obstruction.
The central nervous system weighed 450 g vs. an expected weight of 385 g. Macroscopically, supratentorial cuts showed mild ventricular system dilatation, but there was no presence of hemorrhage of the germinal matrix or any other alterations. Histologically, cuts of the cortex showed normal development with a normal cortical migration for age. There were some hypoxic-appearing neurons with significant retraction of the cytoplasm and hyperchromasia and pyknosis of the nuclei.
The kidneys weighed 16 g vs. an expected weight of 20 g and presented lobulations on the capsular surface, which is normal up to 2 years of age. Seven calyces were counted, which eliminated the diagnosis of renal hypoplasia. According to weight, hypotrophy was evident. There was significant congestion of the papillae at the level of the medulla. Histologically, a normal-appearing cortex was observed. There was no longer glomerulogenesis (the glomeruli were term). There was significant congestion of the vessels located in the glomerular cortex. Upon higher magnification, the latter was able to be confirmed as well as the loss of cortical tubular epithelium secondary to state of shock.
There were no alterations in the esophagus; the gastric mucosa was congested. However, it was histologically intact and there were no contraction bands at the level of the smooth muscles. The small intestine and colon had some areas of mucosal congestion. Histologically, there were areas of congestion in the lamina propia and autolysis. There was presence of ganglionic neurons in the submucosal and myenteric plexus in all cuts. In the cuts of the colon, congestion secondary to shock and contraction and the presence of ganglionic neurons were able to be observed. There were no histological alterations in the pancreas. The acinar cells and islets of Langerhans were without alterations. The spleen weighed 15 g vs. an expected weight of 12 g. Macroscopically, it presented areas of congestion that were histologically corroborated and showed a significant depletion of lymphoid tissue. Bone marrow had normal characteristics with endochondral ossification corresponding to a child the age of this patient and without changes. Nodes did not yet have secondary follicles, only primary follicles. The thymus was observed with some dilated and calcified Hassall corpuscles with mild decrease in lymphoid tissue. The adrenal gland was intact with congestion at the level of the medulla but without hemorrhage. There was interstitial congestion in the testicle. All postmortem cultures were negative.
Based on these findings, diagnoses of cerebral edema, bilateral renal hypotrophy, and multivisceral congestion were made, which is one of the anatomical findings of shock. Long-standing right ventricular hypertrophy was the result of all alterations presented during fetal life. This was reflected in the persistent pulmonary hypertension presented in utero. As an incidental finding, an adrenal gland was found located in the epididymis of the right testicle, which has no clinical significance (Fig. 8).7-10
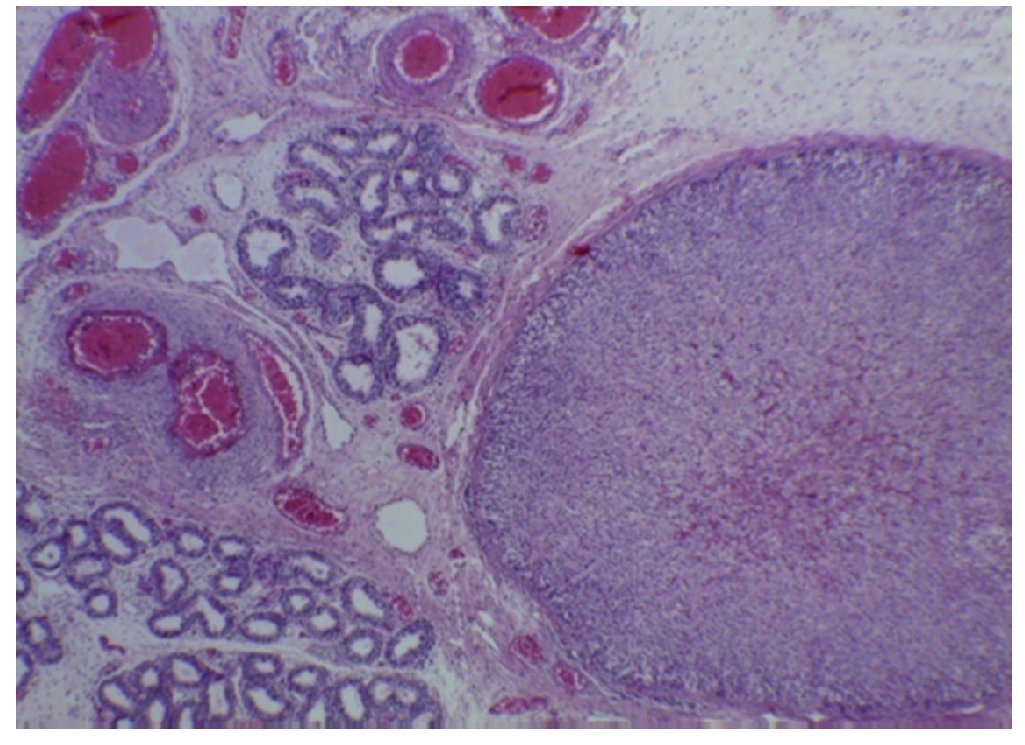
Figure 8 Ectopic adrenal tissue in the right testicle (40×).
5. Final comments
5.1. Neurology (Dr. Saúl Garza Morales)
Fetal medicine has changed and the manner of approach has also changed. One of the themes discussed previously was with respect to the work teams responsible for the newborn and the teams responsible for caring for the fetus, thus underlying the importance of forming interdisciplinary teams with perinatologists, neonatologists, and postnatal surgeons. This is of utmost importance for this and other types of malformations, e.g., urinary tract. When a prenatal patient is being evaluated and treated, other basic studies to be carried out are chromosomal or genetic studies.
5.2. Medical Director (Dr. Jaime Nieto-Zermeño)
Genetic study was performed along with the enthusiastic participation of neonatologists and perinatologists. Prior to fetal surgery, the case is always discussed with Dr. Rogelio Cruz Martínez in Queretaro. Total and unconditional support is provided by Fetal Surgery, Anesthesia and General Pediatric Surgery Groups of the General Hospital of Mexico.
This patient clearly illustrates the possibility of achieving a positive outcome because the initial prognosis was a stillbirth, i.e., the fetus would not have been born. Despite the effort, the patient did not survive. However, a great learning experience was gained.
5.3. Neonatology (Dr. Daniel Ibarra Ríos)
Interdisciplinary management and communication is very important in order to carry out these procedures. From cases with 100% in-utero mortality, one is seeing what can be accomplished and what procedures to offer. However, all areas of care must be under control with the most difficult area being logistics. Fetal surgery cases are reported along with the outstanding work performed with services from nursing, respiratory therapy, materials, anesthesia, etc.
As previously explained, at birth the child is intubated with an open cord. There must be three or four neonatologists who receive the child in a radiant heat incubator. There is a tight relationship among all personnel of the services represented who will gather at the incubator of a seriously ill child. In these children who initially have hydrops, control of hypotension is very difficult. First, the infant need to be hemodynamically stabilized and hypotension and pulmonary vascular hypertension must be controlled. Samples of amniotic fluid must be taken as well as sending the placenta to pathology. On the other hand, the infectious part is very complicated and the necessary experience in regard to related infections is lacking.
The most important procedure is to monitor cardiac frequency and to evaluate the need for chest compressions. Two additional persons should be available for placement of vascular access. There is the opportunity to have access to the umbilical vessels, which allows for venous and arterial monitoring. From there, the patient should be transferred to the NICU and then managed with conventional and high frequency ventilation. Nitric acid, esterase 3 and 5 and rescue vasopressin should be available for management of pulmonary vascular disease. Early recognition of adrenal insufficiency is also important in order that initiation of management with steroids, cortisol measurements, etc. is done when necessary. In summary, it is a very complicated procedure. Lists of comparison are being formalized along with a well-standardized program to see what options can be offered. Intervention from social and psychology services is also important, especially for the support offered to the parents. This study is important because we were able to obtain postmortem findings, which help to acquire experience in these cases.
One component has been the ability to perfect, specifically, intubation times, hypertension management, etc. in order to strengthen the protocol for pulmonary vascular hypertension. These are the patients who were not previously seen because there are some in whom cystic adenomatoid disease is reversed in utero. For this, it is accepted that steroids be administered. The important aspect is to acquire experience in order to drastically change management of these children. This is on its way due to the advent of fetal surgery.
5.4. Pediatric surgery (Dr. Ricardo Ordorica)
Mention was not made of three important services: administrative, genetics and radiology. I believe that the evolution, diagnosis and early treatment allow for more cases to be known. Perinatal medicine is based on which cases should be intervened and which should be allowed to continue to term. The fetus is considered as a patient who has a right to health and treatment, if one exists. In this case, once in the hospital, complete treatment can be offered. This case in particular posed challenges in regard to what is the opportune time for surgical intervention.
5.5. Medical Director (Dr. Jaime Nieto-Zermeño)
In regard to whether a lung transplant was feasible, it was actually impossible. This is a case that provided great learning opportunities.
Ethical disclosures
Protection of human and animal subjects. The authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of data. The authors declare that no patient data appears in this article.
Right to privacy and informed consent. The authors declare that no patient data appears in this article.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 27 May 2015;
accepted 1 June 2015
☆ Please cite this article as: Nieto-Zermeño J, Cruz-Romero EV, Romero-Baizabal BL, Escobar-Sánchez MA. Malformación congénita de la vía aérea tipo I. Bol Med Hosp Infant Mex. 2015. http://dx.doi.org/10.1016/j.bmhimx.2015.06.005
* Corresponding author.
E-mail:bcnhim20091@hotmail.com (M.A. Escobar-Sánchez).