





Polycystic liver disease rarely occurs in isolation as part of autosomal dominant polycystic liver disease, but more commonly, it exists as an extra-renal manifestation of autosomal dominant polycystic kidney disease. The pathogenesis of polycystic liver disease involves defects in the primary cilium of the cholangiocyte, with genetic mutations that impair key proteins integral to the complex functioning of cilia. While most patients are asymptomatic and require no intervention aside from reassurance and genetic counseling, in a minority of patients, polycystic liver disease creates a myriad of symptoms from the compressive effects of enlarged cysts, and can even cause malnutrition and liver decompensation in the severest of cases. In patients with symptomatic disease, a variety of interventional radiology or surgical techniques can be considered, including aspiration with sclerotherapy of a dominant cyst, fenestration, segmental hepatic resection, and even liver transplantation. Although there are no curative medical options for polycystic liver disease, somatostatin analogs hold promise and have shown minimal efficacy in human studies. However, further research is needed to develop more efficacious medical treatments.
Polycystic liver disease (PLD) is an inherited ciliopathy characterized by numerous simple cysts of 1 centimeter or more in diameter within the liver, without evidence for an infectious or traumatic cause. An alternative definition to PLD involves the presence of hepatic cysts occupying at least half of the volume of the hepatic parenchyma. Despite the variation in these definitions in the published literature, most experts would accept the presence of a score or more of simple cysts in the liver to be sufficient to constitute PLD.1
In adults, PLD occurs as a frequent extra-renal manifestation of autosomal dominant polycystic kidney disease (ADPKD), or less commonly, in isolation as autosomal dominant polycystic liver disease (ADPLD). In both scenarios, PLD has a broad spectrum of severity, and in the vast majority of patients, the liver cysts are an incidental finding of no clinical consequence.2,3
The process of cyst formation in both PLD and PKD is as fascinating as it is complex, and cystogenesis in both disorders involves primary defects in cilia lining the biliary epithelium as well as key proteins integral to the functioning of cilia. Given the commonality in the pathogenesis of both PLD and PKD, both disorders fall under the category of hepatorenal fibrocystic diseases or ciliopathies.
This article aims to provide an updated and clinically-oriented summary of our current understanding of PLD. While most patients do not require any therapeutic intervention, mechanical destruction of cysts by either techniques of interventional radiology or surgery remains the mainstay of treatment for the most advanced cases. However, in the near horizon lies the promise of medical treatment options with demonstrated efficacy in animal models and human studies, namely the use of somatostain analogs and mammalian target of rapamysin (mTOR) inhibitors.4
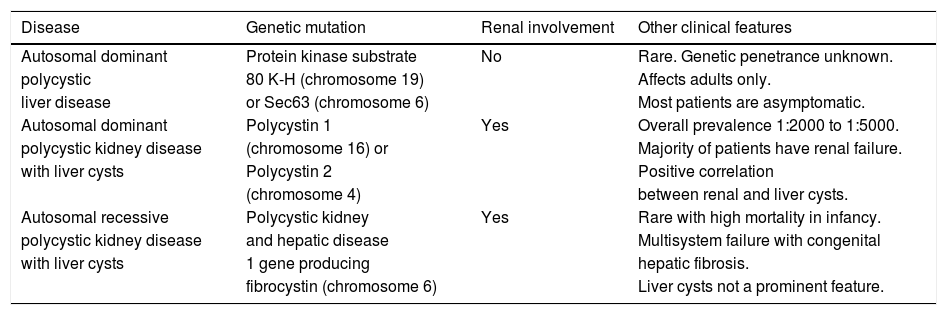
Epidemiology and Genetic AssociationsIn considering the epidemiology of PLD, 3 groups stratified by type of hepatorenal fibrocystic disease must be considered: ADPLD, ADPKD, and autosomal recessive polycystic kidney disease (ARPKD). Salient features of these variants are summarized in table 1.
A summary of polycystic liver diseases.
| Disease | Genetic mutation | Renal involvement | Other clinical features |
|---|---|---|---|
| Autosomal dominant | Protein kinase substrate | No | Rare. Genetic penetrance unknown. |
| polycystic | 80 K-H (chromosome 19) | Affects adults only. | |
| liver disease | or Sec63 (chromosome 6) | Most patients are asymptomatic. | |
| Autosomal dominant | Polycystin 1 | Yes | Overall prevalence 1:2000 to 1:5000. |
| polycystic kidney disease | (chromosome 16) or | Majority of patients have renal failure. | |
| with liver cysts | Polycystin 2 | Positive correlation | |
| (chromosome 4) | between renal and liver cysts. | ||
| Autosomal recessive | Polycystic kidney | Yes | Rare with high mortality in infancy. |
| polycystic kidney disease | and hepatic disease | Multisystem failure with congenital | |
| with liver cysts | 1 gene producing | hepatic fibrosis. | |
| fibrocystin (chromosome 6) | Liver cysts not a prominent feature. |
The prevalence of ADPLD is unknown as there are no robust population-based studies, and published literature estimating the prevalence of ADPLD is likely to underestimate the burden of the disease as most patients are asymptomatic and thus unlikely to ever receive a diagnosis. There is also limited data on the disease’s genetic penetrance.
In contrast to liver cysts associated with PKD, in ADPLD, there is typically no renal involvement.5 While it should be noted that in nearly 80% of patients with presumed ADPLD no genetic mutation is ever identified, in a sizeable minority of patients, a mutation on the short arm of chromosome 19 affecting protein kinase substrate 80 K-H (PRKCSH), that encodes for hepatocystin, or a mutation on the short arm of chromosome 6 affecting Sec63, is found.6-8 Both of these proteins are integral to the functioning of the endoplasmic reticulum in cholangiocytes, and participate in carbohydrate processing, folding and translocation of glycoproteins.7,9,10 However, the precise mechanisms by which PRKCSH and Sec63 promote liver cyst formation have not been fully elucidated.9
Unlike ADPLD, the epidemiology of ADPKD and associated PLD is well-described. ADPKD is the most common monogenetic human renal disease with a prevalence of 1:400-1:1000.11 ADPKD is a heterogeneous disorder that originates from defects in either chromosome 16 (approximately 85% of patients) or chromosome 4 (approximately 15% of patients), thereby mutating the gene products polycystin 1 or polycystin 2, respectively. ADPKD causes renal failure in the majority of affected patients, and liver cysts are the most common extra-renal manifestation. Estimations of the prevalence of PLD in patients with ADPKD range from 20% to greater than 75%.3,12 In a study of 239 patients with ADPKD, Gabow, et al. found that risk factors for the presence of liver cysts include older age, female gender, and history of pregnancy in female patients, particularly a history of multiple pregnancies.13 A strong correlation between advanced renal cysts or renal dysfunction, and liver cysts has also been demonstrated in observational studies.13-15
Contrary to ADPKD, ARPKD is rare, with an incidence of only 1:10,000 to 1:40,000 live births.16,17 ARPKD is associated with a high mortality, and is characterized by congenital hepatic fibrosis, bile duct dilatation, and cyst formation. The disease is caused by mutations of the polycystic kidney and hepatic disease 1 gene (PKHD1) on the short arm of chromosome 6 that encodes fibrocystin (also known as polyductin).15,16 The function of fibrocystin is unknown, but like polycystin 1 and 2, it is contained in the primary cilia of both the kidney and liver, and is implicated in cystogenesis. Unfortunately, ARPKD is associated with a high mortality rate, and many patients die in early life due to respiratory failure from enlarged kidneys, or complications that include hypertension, portal hypertension (secondary to congenital hepatic fibrosis or Caroli’s disease), chronic lung disease, growth retardation, intracranial aneurysms or adrenal insufficiency. While ARPKD is always found in conjunction with congenital hepatic fibrosis, it is less commonly associated with hepatic cystogenesis. Liver cysts are rare in children with ARPKD, although the frequency of liver cysts increases with age.
PathogenesisAs eluded to in the prior section in which the genetics of ADPKD and ADPLD were highlighted, the pathogenesis of hepatic cyst formation in both ciliopathies share similarities as well as differences. Hepatic cyst formation originates from malformation of the ductal plate in the embryonic development of the liver, leading to discrete intralobular bile ductules called von Meyenburg complexes that can be subject to progressive dilatation. In ADPLD, the primary defect involves faulty cell proliferation, whereas in PLD associated with ADPKD, the primary defect is a disruption in cell adhesion.3
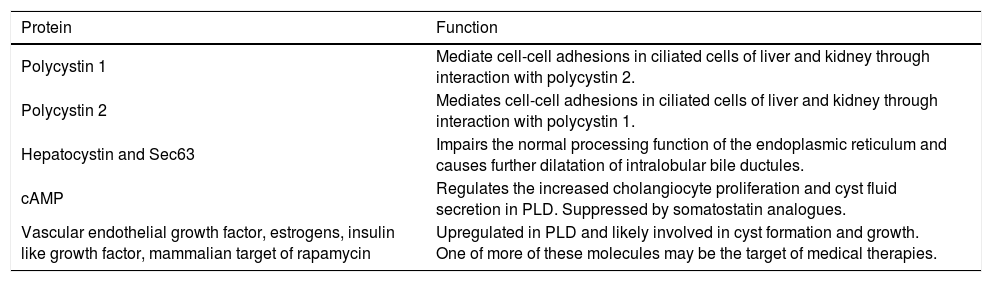
In exploring the pathogenesis of PLD, one must further consider the important function of the polycystin proteins (polycystin 1, polycystin 2, hepatocystin and Sec63). Polycystin-1 interacts with polycystin-2 for functional regulation of both proteins, and collectively the polycystin proteins help mediate cell-cell adhesions in ciliated cells within the liver and kidney.18 In ADPLD, mutated hepatocystin or Sec63 impair the normal processing function of the endoplasmic reticulum and cause further dilatation of intralobular bile ductules, thus advancing cystogenesis.6 A summary of the key proteins involved in the pathogenesis of PLD is found in table 2.
Key functional proteins in polycystic liver disease.
| Protein | Function |
|---|---|
| Polycystin 1 | Mediate cell-cell adhesions in ciliated cells of liver and kidney through interaction with polycystin 2. |
| Polycystin 2 | Mediates cell-cell adhesions in ciliated cells of liver and kidney through interaction with polycystin 1. |
| Hepatocystin and Sec63 | Impairs the normal processing function of the endoplasmic reticulum and causes further dilatation of intralobular bile ductules. |
| cAMP | Regulates the increased cholangiocyte proliferation and cyst fluid secretion in PLD. Suppressed by somatostatin analogues. |
| Vascular endothelial growth factor, estrogens, insulin like growth factor, mammalian target of rapamycin | Upregulated in PLD and likely involved in cyst formation and growth. One of more of these molecules may be the target of medical therapies. |
Cyst formation relies on increased cholangiocyte proliferation and fluid secretion. Cholangiocyte adenosine 3’5’-cyclic monophosphate (cAMP) is the key protein that regulates this process.19 Secretin, a major agonist of cAMP in cholangiocytes, stimulates the insertion of numerous transporters and channels into the apical membrane of cholangiocytes to promote fluid production in hepatic cysts. Suppression of cAMP with somatostain analogs in rat models with ADPKD results in a 22-60% reduction in the size of hepatic cysts, verifying the important role of cAMP is cystogenesis.19 Additional abnormalities involving cholangiocyte proliferation and apoptosis are the subject of intense research in the pathogensis of PLD. Higher levels of vascular endothelial growth factor, estrogens, insulin-like growth factor, and mammalian target of rapamycin (mTOR), among other proteins, are found in abundance in hepatic cysts, and may be the target(s) of further therapies.18,20,21
Diagnosis, Clinical Manifestations and Natural HistoryAs stated, ADPLD is genotypically and phenotypically different than ADPKD. Patients with ADPLD do not have polycystic kidneys, whereas the majority of patients with ADPKD (60% or more) have polycystic livers.12 Overall, the long-term prognosis of ADPLD is benign compared with ADPKD.22
The vast majority of patients with PLD are asymptomatic. A minority of patients will experience symptoms over time correlating with the increase in size of liver cysts. Symptoms predominantly result from the mass effect or compression imposed by massive hepatomegaly from enormous cysts.12 Common symptoms from severe PLD include dyspnea, early satiety, malnutrition, gastroesophageal reflux, back pain, complications from portal hypertension due to inferior vena cava and portal vein compression, or complications from bile duct compression. Symptoms such as abdominal pain can also arise from rupture, torsion, infection or hemorrhage of the hepatic cysts themselves. Rarely, liver failure has been attributed to PLD, but hepatic decompensation is the subject of case reports only.23 Patients with liver cysts associated with PKD are clearly prone to additional symptoms related to renal impairment. A devastating consequence to severe PLD is poor body image, depression, loss of function, and over all poor quality of life.24 Patients with severe PLD are often too debilitated to perform simple tasks involving bending, for example, and this impairs their ability to independently perform activities of daily living. Indeed, patients with severe PLD often describe their condition as equivalent to being 9-months pregnant.
There is sparse robust observational data on the natural history of PLD. The common thinking is that liver cysts in PLD grow slowly over time. This hypothesis is corroborated by several trials published on the PLD population that documents an annual growth rate of hepatic cysts at roughly 0.9 to 3.2%.1,25,26 Risk factors for liver cyst growth include advancing age, female gender, and large renal cyst volume.15 While liver failure and death from isolated PLD is rare, it has been estimated that cystic liver disease accounts for 10% of deaths in patients with ADPKD on HD.15
In the majority of patients with PLD, liver enzymes and liver function tests are within normal or near-normal limits. However, in symptomatic patients, laboratory derangements are much more common. For instance, it has been observed that alkaline phosphatase, gamma-glutamyl transpeptidase, aspartate aminotransferase, and total bilirubin are commonly elevated in patients with advanced disease, at rates anywhere from 15% to 70%.27
Diagnostic imaging is an important tool to confirm the diagnosis of PLD, and help exclude other diseases in the differential diagnosis of hepatomegaly. Abdominal ultrasound and computed tomography (CT) are the first-line imaging modalities of choice for many clinicians due to their excellent ability to image the liver, widespread availability, and acceptable cost, although magnetic resonance can also provide superb data that is particularly useful for operative planning. In PLD, cysts appear smooth-walled and are anechoic on ultrasound. On contrast CT, cysts are non-enhancing and distinct from the hepatic parenchyma (Figure 1). If cysts have irregular thickening, septations, or changes in attenuation or echogenicity, the clinician should consider a complication such as bleeding, infection, or neoplasm (e.g. cystadenomas or cystadenocarcinoma).27 There is no evidence that the asymptomatic patient requires surveillance imaging for monitoring, although clearly imaging has a vital role to play in therapeutic planning in those patients with severe disease who require intervention.
Treatment: Current and Emerging
In addition to the importance of patient education and genetic counseling, there are three general categories for the treatment approach of PLD: conservative management, medical therapy, and surgery (Table 3). The majority of patients, possessing no symptoms or consequences to the presence of liver cysts, will require no treatment whatsoever. In these patients, reassurance and avoidance of repeated abdominal imaging (to limit the cumulative radiation exposure of patients and prevent unnecessary patient anxiety) is appropriate.
Summary of emerging and known treatments for symptomatic polycystic liver disease.
| Treatment category | Treatment |
|---|---|
| Medical | Somatostatin analogues mTOR inhibitors |
| Radiological | Aspiration with sclerotherapy |
| Surgical | Fenestration (laparascopic or open)Segmental hepatic resectionLiver transplantation |
While there are presently no approved medical treatments for PLD, there have been exciting recent advancements with somatostatin analogs.28 In a randomized controlled cross-over study, Caroli, et al. assessed 6 months of treatment with 40 mg of long-acting octreotide debot monthly vs. placebo, and found that the total liver volume decreased by 71 ± 57 mL in the treatment arm, whereas in the placebo group, the total liver volume increased by 14 ± 85 mL; this difference was statistically significant.29 Similarly, in another trial in which patients were randomized in a double-blinded fashion to the somatostatin analog lanreotide at 120 mg monthly versus placebo, investigators found that the total liver volume as measured by CT at week 24 decreased by a mean of 2.9% in the treatment group, whereas the liver volume increased by 1.6% in the placebo group (p = 0.05).26 Likewise, Hogan et al also assessed the efficacy of long-acting octreotide 40 mg monthly versus placebo for one year in 42 patients with severe PLD from ADPKD.25 The primary endpoint of this trial was the change in liver volume as assessed by magnetic resonance between baseline and 1 yr.25 The investigators found that liver volume decreased by 4.95 ± 6.77% (p = 0.048) in the treatment group but was unchanged in the group of patients who received placebo.25 Furthermore, in the patients with ADPKD, the kidney volume was unchanged in the treatment group, but increased in the placebo group by 8.61 ± 10.07% (p = 0.045).25
In the abovementioned trials, there were minimal side effects with somatostatin analogs. Given a consistent demonstration of mild efficacy, these agents may be suitable for therapy in select patients with PLD, although it would appear these agents require continuous use to maintain their effect as demonstrated in a recent open-labeled study.30 While somatostatin analogues would be futile in patients with severe PLD in whom surgery is required, there may be a role for these agents in patients with moderate PLD who are symptomatic but in whom the risks for surgical therapy are not justified, or in whom a surgical intervention is technically challenging.
mTOR inhibitors are another category of medications that could have antiproliferative effects on cysts in polycystic liver and kidney disease in some patients. mTOR is member of the phosphoinositide 3-kinase related kinase (PIKK) family, and is a principal mediator of cell proliferation. In animal models with PKD, mTOR inhibition delays cystogenesis.31 The specific impact of mTOR inhibitors on the growth of kidney cysts, however, is disappointing. For instance, in a randomized controlled trial in which patients received either sirolimus 2 mg daily or placebo for 18 months, the investigators found that there was no difference in kidney cyst growth rates between the two groups.32 Similarly, Walz, et al. determined no prevention of renal cystogenesis, but only retardation of the growth rate of renal cysts, in a group of patients with ADPKD who received everolimus for 2 years versus placebo.33 In contrast, in a study in which kidney transplant recipients with ADPKD and associated PLD were randomized to sirolimus versus tacrolimus, investigators observed a reduction in liver volume in the sirolimus group by 11.9% after a mean treatment time of 19.4 months, as compared with a 14.2% increase in liver volume in the patients who received tacrolimus.20 Although mTOR inhibitors appeared promising in animal models, there is insufficient published data to determine if they will have a potential role in the management of select patients with PLD, particularly given the mixed results of the abovementioned trials.
Radiological and surgical treatmentMechanical removal of hepatic cysts by invasive techniques is the mainstay of treatment for patients with advanced and symptomatic PLD. Possible techniques include cyst aspiration, fenestration, segmental hepatic resection, or liver transplantation.
Aspiration with sclerotherapy is performed under image guidance. The procedure entails aspiration of a dominant cyst, followed by injection of a sclerosing agent that causes destruction of the epithelial lining of the cyst wall, thereby impairing fluid generation. Aspiration is indicated for a large cyst of at least 5 centimeters in diameter that is believed to contribute to symptoms.1 Examples of possible sclerosing agents include ethanol, ethanolamine oleate, minocycline or tetracycline; these compound destroy the cyst wall by creating a low pH within the cyst.1,34 Although a single session is typically sufficient for treatment of a dominant cyst, some patients will occasionally require more than one session for cyst obliteration.35 Aspiration with sclerotherapy has an excellent safety profile, although complications of pain secondary to peritoneal irritation have been reported. Long-term outcomes include regression, partial regression, and recurrence, all of which occur at a frequency of approximately 20%.1 However, the majority of patients who undergo dominant cyst aspiration are asymptomatic following the procedure.1
Unlike aspiration, fenestration is preferred in patients who have multiple cysts in close proximity, but away from cranial segments of the liver that are difficult to access by this technique.36 Fenestration involves both aspiration and surgical unroofing. While it is usually done laprascopically, conversion to open technique is sometimes required.36 With respect to clinical outcomes, the vast majority of patients enjoy immediate resolution of symptoms, whereas up to a quarter of patients ultimately develop recurrence of cysts and symptoms.1 Complications of fenestration include ascites, pleural effusion, hemorrhage and bile leakage.37 Overall, 23% and 2% of patients have morbidity or mortality from fenestration, respectively.1 Published data on causes of death following fenestration include shock, hepatic abscess and renal failure.1
Segmental hepatic resection is another surgical approach to advanced PLD that is specifically considered for patients with massive hepatomegaly but at least 1 segment with normal or near-normal liver parenchyma.37 Segmental resection for this indication is not widely performed, and there are very few centers of expertise. A hepatobiliary surgeon would ideally attempt to retain at least 30% of the remnant liver.38 In select cases, a combination of segmental resection with fenestration of remaining cysts can be considered.
Liver transplantation is the only curative treatment for those patients with severe PLD in whom partial resection with or without fenestration is unsuitable. There are no generic indications to list a patient with PLD for liver transplantation as hepatic functioning is usually normal. Instead, clinical decision-making rests on the careful consideration of a multidisciplinary panel. Patients who suffer tremendous morbidity or otherwise incurable medical complications, and who are not deemed candidates for less invasive surgical techniques by an expert hepatobiliary surgeon, should be assessed on a case-by-case basis. Owing to the uniqueness of liver transplantation for PLD, many organ allocation systems provide exception points for listing patients following approval by a regional review board.39
For those patients who ultimately undergo liver transplantation or, in the setting of advanced polycystic kidney disease, combined liver and kidney transplantation, long-term clinical outcomes are superb. For instance, the 1-and 5-year patient survival for hepatic transplantation alone for PLD approximate 90%, respectively, and 1-and 5-year survival rates after combined transplantation approximate 80-85%, respectively.1,40 Patients undergoing combined liver/kidney transplantation from the same organ donor have superior outcomes as compared to patients receiving the grafts sequentially from two separate donors due to a reduction in renal rejection in the former approach.41 As such, concurrent combined liver and kidney transplantation from one donor should be the standard-of-care for patients with severe polycystic liver and kidney disease with advanced renal insufficiency or requiring renal replacement therapy.
In among the largest single-center experiences published in North America, we reported on the long-term outcomes following transplantation in 15 subjects, 14 of whom had liver transplantation alone, and 10 of whom had associated ADPKD, for severe PLD.42 We observed a higher rate of vascular complications, with 3 of 15 (20%) patients experiencing early hepatic artery thrombosis.42 We posit that vascular complications may be more frequent in patients transplanted for PLD due to the lack of intrinsic coagulopathy in the operative period (in contrast to patients with end-stage liver disease), and the excess space in the abdominal cavity after hepatectomy of the enlarged organ which can theoretically increase the risk of torsion or volvulus of the hepatic graft, and thus lead to thrombosis. Our finding that the majority of patients with PLD from ADPKD do not require concurrent kidney transplantation concurs with the single-center analysis of Piernne, et al., who similarly observed outstanding long-term patient and graft survival rates, and satisfactory long-term renal function, following liver transplantation for PLD.43
There is evidence that liver transplantation improves the quality of life of those with advanced PLD. Kirchner, et al. assessed a cohort of patients who underwent liver transplantation (n = 21) or combined liver/kidney transplantation (n = 15) for cystic disease at a single-center in Germany from 1990 to 2006.24 After a mean of 62 months of follow-up, the authors found that 91% of patients had a substantial improvement in health-related quality of life measures as assessed by the standard short-form 36 and a self-designed quality-of-life questionnaire.24
ConclusionPLD is a fascinating condition that occurs both in isolation as part of ADPLD, or, more commonly, as an extra-renal manifestation of ADPKD. This review highlighted available evidence on the epidemiology, genetics, pathogenesis, clinical manifestations, natural history and treatment of PLD. It should be re-emphasized that the vast majority of patients with PLD require no medical or surgical intervention or follow-up with respect to liver cysts, but clearly benefit from genetic counseling and education. In contrast, those patients with advanced liver cysts suffer from a myriad of symptoms leading to significant morbidity. A variety of minimally invasive interventional radiology techniques and surgical options are available for patients with severe polycystic disease, and each procedure has specific indications, contraindications and potential complications based on the size and location of the cyst(s), as well as local expertise. In the severest of cases, liver transplantation or combined liver and kidney transplantation, should be considered, and well-selected patients enjoy outstanding long-term clinical outcomes and an improvement in their quality of life. Exciting advancements are being made in medical approaches to PLD, although more progress is needed in that area.
Conflict of Interest or Financial DisclosuresNone.



