







Genetic background may be involved in the mechanisms of liver injury and the development of non-alcoholic fatty liver disease (NAFLD). However, its contributions to the long-term outcome of NAFLD have been unclear.
MethodsWe enrolled 314 Japanese patients with biopsy-confirmed NAFLD from 2000 to 2018 (161 men [51.3%]; median age, 53 [14–84] years; 114 with advanced fibrosis [37.5%]) in the patients without hepatocellular carcinoma at diagnosis. Genomic DNA was extracted from peripheral blood and single nucleotide polymorphisms (SNPs) were analyzed. Associations of mortality with patatin-like phospholipase 3 (PNPLA3) and aldehyde dehydrogenase 2 (ALDH2) were analyzed. Finally, a subgroup analysis according to lifestyle-related disease was performed.
ResultsDuring the median 7 years of follow-up, 20 patients (6.4%) died (13 liver-related [4.1%] and 7 non-liver-related deaths [2.2%]). Patients with ALDH2 (non-GG genotype) who had reduced alcohol metabolism tended to have a poor prognosis (p = 0.06). Patients carrying both risk SNPs of PNPLA3 (GG) and ALDH2 (non-GG) had a significantly poor prognosis (p = 0.01). In the subgroup analysis, patients with PNPLA3 (GG) who were non-diabetics (p = 0.06) or non-dyslipidemic (p = 0.03), with ALDH2 (non-GG) who were non-dyslipidemic (p = 0.01) or hypertensive (p = 0.03), also had a poor prognosis. The Cox analysis revealed that ALDH2 (non-GG) was associated with a poor prognosis (Hazard ratio: 4.568, 95% Confidence Interval: 1.294–16.131, p = 0.02) similar to the liver function tests.
ConclusionsGenetic background may affect NAFLD prognosis and ALDH2 SNP could predict the outcome.
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent cause of chronic liver disease, as demonstrated by a Japanese cohort study, in which 29.7% of patients with a chronic liver disease had NAFLD [1]. Lifestyle-related diseases such as diabetes, dyslipidemia, and hypertension are causes of NAFLD. Most patients with NAFLD have obesity and/or lifestyle-related diseases; however, some lean patients develop NAFLD [2]. Genetic background is thought to influence the development of NAFLD/nonalcoholic steatohepatitis in Japanese people. A risk allele of the patatin-like phospholipase 3 (PNPLA3) single nucleotide polymorphism (SNP; rs738409 [G], encoding I148M) is more frequent in Japan than in Western countries (GG 21% in Japan and 5% in Western countries) [3–5]. The frequency of the GG genotype is increased in patients with NAFLD (GG 37% in Japan and 12% in Western countries) [6,7]. It is localized in the endoplasmic reticulum and on the surface of lipid droplets [8]. The PNPLA3 I148 M mutation influences intrahepatic remodeling and reduces very-low-density lipoprotein secretion [9]. This mutation disrupts ubiquitylation and proteasomal degradation of PNPLA3, which impairs mobilization of triglycerides from lipid droplets inducing NAFLD [10]. Thus, a risk allele in PNPLA3 is strongly associated with increased hepatic fat levels and hepatic inflammation [11]. We previously reported that the PNPLA3 GG genotype is associated with the initiation and development of fibrosis in patients with NAFLD [12]. However, its association with prognosis and lifestyle-related diseases in patients with NAFLD is unclear.
To compare the associations of genetics with alcoholic liver disease, we evaluated the genotype of aldehyde dehydrogenase 2 (ALDH2). ALDH2 is involved in alcohol metabolism, aldehyde detoxification [13], and liver protection [14]. The cellular concentration of highly cytotoxic aldehydes is regulated by the balance between the various alcohol dehydrogenase and aldehyde dehydrogenase isoforms [13]. Additionally, ALDH2 is involved lipid peroxidation [15]. Our previous findings suggested that the ALDH2 GG genotype (more rapid aldehyde metabolism) was present at a lower frequency in patients with NAFLD, compared to patients with alcoholic liver disease [12]. In contrast, the AA/AG genotypes of ALDH2, which are linked to suppression of aldehyde metabolism, were detected at a high frequency in patients with NAFLD. The ALDH2 AA genotype was related to alcohol flushing syndrome and esophageal or pharyngeal cancer [16,17]. However, the associations of these genotypes with the onset and outcome of NAFLD are unclear. Here, we investigated the contributions of genetic background and lifestyle-related diseases to mortality in Japanese patients with NAFLD.
2Methods2.1Patients and study designThis was an observational single-center study at Tokyo Women’s Medical University Hospital. From 2000 to 2018, 314 Japanese patients with biopsy-confirmed or clinically diagnosed NAFLD were enrolled in this study. The diagnosis of NAFLD was based on the following criteria [18]: (1) >5% macrovesicular steatosis of hepatocytes or hepatic steatosis by an imaging modality; (2) intake of <20 g of ethanol/day in women and <30 g/day in men; and (3) appropriate exclusion of drug-induced steatohepatitis. The pathological stage of NAFLD was evaluated in accordance with the classification system described by Brunt et al. [19]. Fibrosis staging was based on the location and extent of fibrosis, as follows: stage 1 (F1), zone 3 perisinusoidal fibrosis; stage 2 (F2), portal fibrosis; stage 3 (F3), bridging fibrosis in addition to portal fibrosis; and stage 4 (F4), cirrhosis.
Detailed clinical and demographic data were collected, including age, sex, body mass index (BMI), and lifestyle-related diseases. Patients with BMI ≥ 25 kg/m2 fit the criteria for obesity. Diagnoses of diabetes mellitus, dyslipidemia, and hypertension were based on the criteria for metabolic syndrome [20,21] or use of medication. Diabetes was diagnosed by random blood glucose >200 mg/dL or fasting blood sugar (FBS) >126 mg/dL or hemoglobin A1c (HbA1c) ≥ 6.5% National Glycohemoglobin Standardization Program (NGSP). Dyslipidemia was diagnosed based on elevated total cholesterol levels (>220 mg/dL) and/or triglycerides (>150 mg/dL). Hypertension was diagnosed if patients had a blood pressure of ≥140/90 mmHg.
Laboratory data were collected at the time of liver biopsy. Homeostatic model assessment for insulin resistance was calculated from the glucose and insulin levels, to quantify insulin resistance and β-cell function [22]. The FIB-4 index was defined as: age [years] × aspartate aminotransferase [IU/L]) ÷ (platelet count [109/L] × √alanine aminotransferase [IU/L]) [23].
Hepatocellular carcinoma (HCC) was diagnosed histologically or based on imaging findings consistent with the diagnosis, using at least two of the following modalities, in accordance with clinical guidelines: abdominal ultrasound, computed tomography, and/or magnetic resonance imaging [24,25]. The patients diagnosed with HCC were excluded.
Patients with NAFLD were followed up at 2–3-month intervals at the outpatient clinic to assess lifestyle modifications, including diet and physical activity level, as well as for treatment of associated metabolic disorders (diabetes, dyslipidemia, and hypertension). Imaging analyses were performed at 6–12-month intervals to screen for HCC.
This study was conducted in accordance with the principles of the Declaration of Helsinki and the ethical guidelines of the Tokyo Women’s Medical University Hospital (Tokyo, Japan). The Institutional Review Board of Tokyo Women’s Medical University Hospital approved the study protocol. Informed consent was obtained from all participants.
2.2Analysis of genetic polymorphismsGenomic DNA was extracted from whole blood. rs738409 (PNPLA3, n = 309) and rs671 (ALDH2 family; ALDH2, n = 314) were assessed using the MassARRAY® System (Agena Bioscience, San Diego, CA, USA), with iPLEX® chemistry, which combines mass spectrometry with polymerase chain reaction. The primers were designed against specific sites in each gene. The primer sequences (5′–3′) were as follows: rs671_W1_F: ACGTTGGATGTTGGTGGCTACAAGATGTCG; rs671_W1_R: ACGTTGGATGAGGTCCCACACTCACAGTTT; rs671_W1_E-A: CACACTCACAGTTTTCACTT; rs738409_W1_F: ACGTTGGATGAGAGAAAGCCGACTTACCAC; rs738409_W1_R: ACGTTGGATGTCACAGGCCTTGGTATGTTC; rs738409_W1_E-A: GGTATGTTCCTGCTTCAT.
2.3Statistical analysisData are presented as medians and ranges. Differences were assessed by the Mann–Whitney U test or χ2 test using SPSS software (SPSS Inc., Chicago, IL, USA); differences with p < 0.05 were considered statistically significant. Cumulative survival curves were constructed by the Kaplan–Meier method according to the presence of obesity (BMI ≥ 25 kg/m2), diabetes, dyslipidemia, and hypertension. Statistical significance of differences in survival was evaluated by the log-rank test.
We investigated the relationships of the following factors with mortality by multivariate analysis: age, fibrosis stage, serum levels of albumin (g/dL), platelet count (×104/μL), prothrombin time (%), FIB-4 index, and SNPs of PNPLA3 and ALDH2. The Cox proportional hazard model was used to assess the hazard ratios (HR) and 95% confidence intervals (CI) for a risk evaluation of overall survival.
3Results3.1Demographics and survival rate of patients with NAFLDFrom 2000 to 2018, 314 Japanese patients with biopsy-confirmed or clinically diagnosed NAFLD were enrolled in the study (161 men [51.3%]; median age, 53 years [range: 14–84 years]; 114 with advanced fibrosis [37.5%]). Table 1 lists the patient demographics at the time of liver biopsy. All patients had chronic liver diseases. The incidences of type 2 diabetes (n = 155, 49.4%), dyslipidemia (n = 214, 68.2%), and hypertension (n = 163, 51.9%) at the time of liver biopsy were recorded. Some patients had no metabolic diseases or obesity (n = 15); others had one (n = 63), two (n = 82), and three comorbidities (n = 101), respectively; some (n = 53) had four complications. There was no significant difference between the PNPLA3 or ALDH2 genotypes. Fig. 1 presents the total survival rate by Kaplan–Meier analysis (Fig. 1a). During the median 7 years of follow-up, 20 patients (6.4%) died (13 liver-related [4.1%] and 7 non-liver-related deaths [2.2%]). Nine new HCC cases were observed. There was no significant difference in outcome by the numbers of complicating metabolic factors.
Characteristics of patients with NAFLD according to risk SNPs.
| Variable | Total (PNPLA3 n = 309; ALDH2 n = 314) | PNPLA3 GG genotype (n = 131) | PNPLA3 non-GG genotype (n = 178) | PNPLA3 p-value | ALDH2 GG genotype (n = 154) | ALDH2 non-GG genotype (n = 160) | ALDH2 p-value |
|---|---|---|---|---|---|---|---|
| Age (years) | 53 (14–84) | 55 (14–83) | 49 (15–79) | <0.01 | 53 (18–84) | 53 (14–79) | 0.91 |
| Male sex (%) | 161 (51.3%) | 62 (47.3%) | 98 (55.1%) | 0.18 | 71 (46.1%) | 90 (56.3%) | 0.07 |
| BMI (kg/m2) | 27.1 (12.8–46.0) | 26.8 (16.1–46.0) | 27.4 (12.8–45.0) | 0.49 | 27.3 (12.8–46.0) | 27.1 (16.1–43.7) | 0.71 |
| Obesity (BMI ≥ 25 kg/m2) | 210 (66.9%) | 86 (65.6%) | 120 (67.4%) | 0.74 | 102 (66.2%) | 108 (67.5%) | 0.81 |
| Diabetes (%) | 155 (49.4%) | 66 (50.4%) | 87 (48.9%) | 0.79 | 72 (46.8%) | 83 (51.9%) | 0.36 |
| Dyslipidemia (%) | 214 (68.2%) | 88 (67.2%) | 122 (68.5%) | 0.80 | 102 (66.2%) | 112 (70.0%) | 0.47 |
| Hypertension (%) | 163 (51.9%) | 74 (56.5%) | 86 (48.3%) | 0.46 | 73 (47.4%) | 90 (56.3%) | 0.12 |
| Fibrosis (%) | 0.03 | 0.55 | |||||
| Mild; F0–2 | 190 (62.5%) | 71 (55.0%) | 115 (67.6%) | 95 (64.2%) | 95 (60.9%) | ||
| Advanced; F3–4 | 114 (37.5%) | 58 (45.0%) | 55 (32.4%) | 53 (35.8%) | 61 (39.1%) | ||
| Laboratory data | |||||||
| Albumin (g/dL) | 4.4 (2.3–5.5) | 4.3 (2.3–5.4) | 4.5 (2.7–5.5) | <0.01 | 4.4 (2.7–5.3) | 4.4 (2.3–5.5) | 0.91 |
| Total bilirubin (mg/dL) | 0.7 (0.2–6.6) | 0.8 (0.2–2.9) | 0.7 (0.3–6.1) | 0.58 | 0.7 (0.2–3.9) | 0.7 (0.3–6.6) | 0.27 |
| Aspartate aminotransferase (U/L) | 51 (13–351) | 52 (19–351) | 48 (13–231) | 0.50 | 51 (13–351) | 51 (15–231) | 0.76 |
| Alanine transaminase (U/L) | 75 (9–911) | 74 (16–911) | 75 (9–483) | 1.00 | 71 (9–911) | 80 (13–373) | 0.86 |
| γ-Glutamyltransferase (U/L) | 64 (13–849) | 70 (13–665) | 62 (15–849) | 0.75 | 66 (13–849) | 63 (18–665) | 0.55 |
| Fasting blood glucose (mg/dL) | 101 (59–433) | 100 (74–433) | 103 (59–252) | 0.56 | 102 (59–433) | 100 (75–258) | 0.48 |
| Hemoglobin A1c (%) | 5.8 (3.7–12.0) | 5.9 (3.9–11.3) | 5.8 (3.7–12.0) | 0.44 | 5.8 (3.7–10.5) | 5.9 (3.9–12.0) | 0.48 |
| IRI (μU/mL) | 11.5 (1.3–204.9) | 11.2 (1.6–204.9) | 11.5 (1.3–166.8) | 0.40 | 11.1 (1.3–204.9) | 11.9 (1.8–166.8) | 0.41 |
| HOMA-IR | 2.92 (0.55–51.60) | 2.93 (1.00–51.60) | 2.92 (0.75–42.40) | 0.20 | 2.78 (0.55–51.60) | 3.06 (0.88–42.40) | 0.40 |
| Triglycerides (mg/dL) | 137 (37–674) | 127 (37–415) | 141 (41–674) | 0.15 | 132 (37–674) | 140 (39–593) | 0.42 |
| Total cholesterol (mg/mL) | 198 (67–482) | 201 (81–328) | 198 (67–482) | 0.19 | 206 (67–482) | 194 (94–328) | 0.17 |
| Ferritin (ng/mL) | 223 (4–2674) | 225 (10–2674) | 217 (4–1782) | 0.32 | 224 (4–1782) | 217 (10–2674) | 0.77 |
| Platelet counts (×104/μL) | 20.4 (3.4–96.0) | 20.1 (3.7–96.0) | 21.3 (4.8–44.4) | 0.08 | 21.9 (4.8–44.4) | 20.1 (3.4–96.0) | 0.63 |
| Prothrombin time (%) | 95.8 (11.8–110.0) | 91.0 (11.8–110.0) | 98.2 (12.8–100.0) | 0.04 | 96.1 (12.8–110.0) | 93.8 (11.8–100.0) | 0.53 |
| AFP (ng/mL) | 3 (1–157) | 3 (1–157) | 3 (1–12) | 0.19 | 3 (1–14) | 3 (1–157) | 0.36 |
| FIB-4 index | 1.38 | 1.73 | 1.25 | <0.01 | 1.35 | 1.45 | 0.25 |
| (0.23–23.41) | (0.26–10.03) | (0.23–9.63) | (0.35–9.63) | (0.23–23.41) |
NAFLD, non-alcoholic fatty liver disease; SNPs, single nucleotide polymorphisms; PNPLA3, patatin-like phospholipase 3; ALDH2, aldehyde dehydrogenase 2; BMI, body mass index; IRI, immunoreactive insulin; HOMA-IR, homeostatic model assessment for insulin resistance, AFP, alpha-fetoprotein; FIB-4, fibrosis-4.
 and genetic background (PNPLA3, b; ALDH2, c; PNPLA3 + ALDH2, d) groups. Patients with the ALDH2 GG genotype tended to have a higher survival rate than patients with the non-GG genotype (p = 0.06, c). Patients with both risk SNPs of PNPLA3 GG and ALDH2 non-GG genotype had a significantly poorer prognosis (p = 0.01, d). NAFLD, non-alcoholic fatty liver disease; PNPLA3, patatin-like phospholipase 3; ALDH2, aldehyde dehydrogenase 2.")
Overall mortality of patients with NAFLD. Survival rates were evaluated by Kaplan–Meier analysis in the overall (a) and genetic background (PNPLA3, b; ALDH2, c; PNPLA3 + ALDH2, d) groups.
Patients with the ALDH2 GG genotype tended to have a higher survival rate than patients with the non-GG genotype (p = 0.06, c). Patients with both risk SNPs of PNPLA3 GG and ALDH2 non-GG genotype had a significantly poorer prognosis (p = 0.01, d).
NAFLD, non-alcoholic fatty liver disease; PNPLA3, patatin-like phospholipase 3; ALDH2, aldehyde dehydrogenase 2.
The survival rate did not differ significantly differ between the PNPLA3 GG and non-GG genotypes (p = 0.34, Fig. 1b). Patients with the ALDH2 GG genotype tended to have a higher survival rate, compared to patients with the non-GG genotype (p = 0.06, Fig. 1c). Patients with both risk SNPs of PNPLA3 GG and ALDH2 non-GG genotype had a significantly poorer prognosis (p = 0.01, Fig. 1d).
3.2Patient profile according to genetic backgroundPatients with the PNPLA3 GG genotype tended to be significantly older and were more likely to have advanced fibrosis than patients with the non-GG genotype (p < 0.01, p = 0.03, respectively). Serum levels of albumin were significantly lower and platelet counts tended to be lower in patients with the GG genotype of PNPLA3 than in patients with the non-GG genotype (PNPLA3 GG vs. non-GG, albumin: 4.3 vs. 4.5 g/dL, p < 0.01 and platelet counts: 20.1 vs. 21.3 × 104/μL, p = 0.08). The prothrombin time (%) was significantly lower and the FIB-4 index was significantly higher in patients with the GG genotype (prothrombin time: 91.0 vs. 98.2 %, p = 0.04 and FIB-4 index: 1.73 vs. 1.25, p < 0.01). Ten patients with PNPLA3 GG died (liver-related death in 7 [3 cases due to HCC or intrahepatic cholangiocarcinoma and the others were due to liver failure] and non-liver-related death in 3 [2 cases of disseminated intravascular coagulation due to infection and 1 due to ovarian cancer]); 9 patients with the non-GG genotype died (liver-related death in 5 [2 cases due to HCC, 1 case of varix bleeding, and 2 cases of liver failure] and non-liver-related death in 4 [1 case each of gastric cancer, duodenal cancer, suicide and unknown]). The mortality did not differ after age and fibrosis status were matched with the conditions.
In ALDH2 analysis, there were more male patients with the ALDH2 non-GG genotype than patients with the GG genotype (GG 46.1%, non-GG 56.3%, p = 0.07). The biochemical results did not differ significantly between the patients with ALDH2 GG and non-GG genotypes. Among patients with the ALDH2 non-GG genotype, 10 died of liver-related causes, 2 died of unknown causes with suspected coronary disease, and 4 died of infection or malignancy without HCC. Of the patients with the ALDH2 GG genotype, 6 died (liver-related and non-liver related deaths: one case of varix bleeding and 2 cases of liver failure; 1 case each of gastric cancer, duodenal cancer, and disseminated intravascular coagulation). The fibrosis status did not significantly differ between patients with ALDH2 GG and non-GG genotypes.
3.3Survival rate according to PNPLA3/ALDH2 in the lifestyle-related diseases subgroupNext, we compared patients with and without the risk SNP of PNPLA3 (GG vs. non-GG). To exclude any effect on survival rates, we performed a subgroup analysis according to lifestyle-related diseases (Fig. 2). The survival rates were not significantly different in patients with and without the risk SNP of PNPLA3 according to obesity (BMI < 25: p = 0.10, Fig. 2a; BMI ≥ 25: p = 0.62, Fig. 2b), diabetes (p = 0.53, Fig. 2d), and dyslipidemia (p = 0.49, Fig. 2f). In patients without diabetes, those with the PNPLA3 GG genotype tended to have a poor prognosis (p = 0.06, Fig. 2c). Median BMI in patients without diabetes and with the PNPLA3 GG genotype was 27.3 kg/m2. In contrast, it was 27.6 kg/m2 in patients of the non-GG genotype. There was no significant difference between the PNPLA3 genotypes. In patients without dyslipidemia, those with the PNPLA3 GG genotype had a significantly lower survival rate (p = 0.03, Fig. 2e). The relationship with survival was not statistically significant in patients with and without hypertension (non-hypertension: p = 0.31, Fig. 2g; hypertension: p = 0.70, Fig. 2h).
 BMI < 25 kg/m2, (b) BMI ≥ 25 kg/m2, (c) non-diabetes, (d) diabetes, (e) non-dyslipidemia, (f) dyslipidemia, (g) non-hypertension, and (h) hypertension subgroups. The survival rate did not differ significantly among patients with and without obesity (BMI < 25 kg/m2: p = 0.10, a; BMI ≥ 25 kg/m2: p = 0.62, b) and hypertension (non- hypertension: p = 0.31, g; hypertension: p = 0.70, h). In patients without diabetes or dyslipidemia, the GG genotype was associated with a poorer survival rate in non-diabetic patients (p = 0.06, c) and a significantly poorer survival rate in non-dyslipidemia (p = 0.03, e). The relationship was not statistically significant in patients with diabetes or dyslipidemia (diabetes: p = 0.53, d; dyslipidemia: p = 0.49, f). PNPLA3, patatin-like phospholipase 3; SNP, single nucleotide polymorphism; BMI, body mass index; DM, diabetes mellitus; DL, dyslipidemia; HT, hypertension.")
Survival rates of PNPLA3 SNPs in the complications of lifestyle-related disease subgroups.
Survival rates were evaluated by Kaplan–Meier analysis in the (a) BMI < 25 kg/m2, (b) BMI ≥ 25 kg/m2, (c) non-diabetes, (d) diabetes, (e) non-dyslipidemia, (f) dyslipidemia, (g) non-hypertension, and (h) hypertension subgroups.
The survival rate did not differ significantly among patients with and without obesity (BMI < 25 kg/m2: p = 0.10, a; BMI ≥ 25 kg/m2: p = 0.62, b) and hypertension (non- hypertension: p = 0.31, g; hypertension: p = 0.70, h). In patients without diabetes or dyslipidemia, the GG genotype was associated with a poorer survival rate in non-diabetic patients (p = 0.06, c) and a significantly poorer survival rate in non-dyslipidemia (p = 0.03, e). The relationship was not statistically significant in patients with diabetes or dyslipidemia (diabetes: p = 0.53, d; dyslipidemia: p = 0.49, f).
PNPLA3, patatin-like phospholipase 3; SNP, single nucleotide polymorphism; BMI, body mass index; DM, diabetes mellitus; DL, dyslipidemia; HT, hypertension.
In ALDH2 analysis, the relationship with survival was not statistically significant between patients with ALDH2 GG and non-GG genotypes according to obesity (BMI < 25: p = 0.17, Fig. 3a; BMI ≥ 25: p = 0.12, Fig. 3b) and diabetes (non-diabetes: p = 0.17, Fig. 3c; diabetes: p = 0.25, Fig. 3d). Survival rates of ALDH2 non-GG were significantly worse in patients without dyslipidemia (non-dyslipidemia: p = 0.01; Fig. 3e, dyslipidemia: p = 0.76, Fig. 3f) and those with hypertension (non-hypertension: p = 0.89, Fig. 3g; hypertension: p = 0.03, Fig. 3h).
 BMI < 25 kg/m2, (b) BMI ≥ 25 kg/m2, (c) non-diabetes, (d) diabetes, (e) non-dyslipidemia, (f) dyslipidemia, (g) non-hypertension, and (h) hypertension subgroups. The survival rate did not differ significantly among patients with obesity (BMI < 25 kg/m2: p = 0.17, a; BMI ≥ 25 kg/m2: p = 0.12, b) and diabetes (non- diabetes: p = 0.17, c; diabetes: p = 0.25, d). The survival rate of the non-GG genotype of ALDH2 was significantly lower in patients without dyslipidemia and with hypertension (non-dyslipidemia p = 0.01, e; hypertension p = 0.03, h). The relationship was not statistically significant in patients with dyslipidemia or without hypertension (dyslipidemia: p = 0.76, f; non-hypertension: p = 0.89, g). ALDH2, aldehyde dehydrogenase 2; SNP, single nucleotide polymorphism; BMI, body mass index DM, diabetes mellitus; DL, dyslipidemia; HT, hypertension.")
Survival rates of ALDH2 SNPs in the complications of lifestyle-related disease subgroups.
Survival rates were evaluated by Kaplan–Meier analysis in the (a) BMI < 25 kg/m2, (b) BMI ≥ 25 kg/m2, (c) non-diabetes, (d) diabetes, (e) non-dyslipidemia, (f) dyslipidemia, (g) non-hypertension, and (h) hypertension subgroups.
The survival rate did not differ significantly among patients with obesity (BMI < 25 kg/m2: p = 0.17, a; BMI ≥ 25 kg/m2: p = 0.12, b) and diabetes (non- diabetes: p = 0.17, c; diabetes: p = 0.25, d). The survival rate of the non-GG genotype of ALDH2 was significantly lower in patients without dyslipidemia and with hypertension (non-dyslipidemia p = 0.01, e; hypertension p = 0.03, h). The relationship was not statistically significant in patients with dyslipidemia or without hypertension (dyslipidemia: p = 0.76, f; non-hypertension: p = 0.89, g).
ALDH2, aldehyde dehydrogenase 2; SNP, single nucleotide polymorphism; BMI, body mass index DM, diabetes mellitus; DL, dyslipidemia; HT, hypertension.
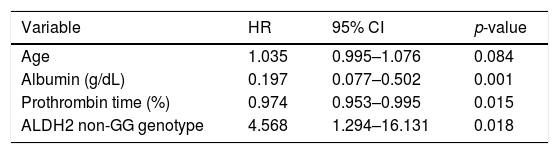
We investigated the factors related to mortality by Cox multivariate analysis (age, fibrosis stage, serum levels of albumin, platelet count, prothrombin time, FIB-4 index, and SNPs of PNPLA3 and ALDH2, Table 2). In addition to the serum markers reflecting liver function such as decreased serum albumin level and prothrombin time (%), the ALDH2 non-GG genotype was found to be significantly related to mortality (HR: 4.568, 95% CI: 1.294–16.131, p = 0.018).
Factors related to mortality by the Cox proportional hazard model for multivariate analysis.
| Variable | HR | 95% CI | p-value |
|---|---|---|---|
| Age | 1.035 | 0.995–1.076 | 0.084 |
| Albumin (g/dL) | 0.197 | 0.077–0.502 | 0.001 |
| Prothrombin time (%) | 0.974 | 0.953–0.995 | 0.015 |
| ALDH2 non-GG genotype | 4.568 | 1.294–16.131 | 0.018 |
ALDH2, aldehyde dehydrogenase 2, HR, hazard ratio; 95% CI, Confidence Interval; SNPs, single nucleotide polymorphisms; PNPLA3, patatin-like phospholipase 3.
Factors; age; fibrosis stage; serum levels of albumin (g/dL), platelet count (×104/μL); prothrombin time (%); Fibrosis-4 index and SNPs of PNPLA3 and ALDH2.
We assessed whether genetic background contributed to mortality in Japanese patients with NAFLD and performed a subgroup analysis according to lifestyle-related diseases. Although the overall mortality rate did not differ significantly in patients with PNPLA3 GG and non-GG genotypes, in the patients without comorbidities of diabetes or dyslipidemia and those carrying the risk SNP had a poor prognosis. The survival rate was significantly lower in patients with the ALDH2 non-GG genotype, especially in those without dyslipidemia. Therefore, the effect of genetic background might be greater in patients with NAFLD who do not have dyslipidemia.
In this study, 20 patients (6.4%) died (13 liver-related [4.1%] and 7 non-liver-related [2.2%] deaths) during the median 7 years of follow-up. The PNPLA3 GG genotype has been associated with the histological severity of NAFLD in a meta-analysis [26]. We previously reported that the GG or CG genotypes of PNPLA3 were significantly more frequent in patients with liver cirrhosis than in patients without cirrhosis (p < 0.05) [12]. The PNPLA3 I148M SNP is associated with an increased liver disease mortality rate in the United States population [27]. However, PNPLA3 was reported not to affect overall mortality [28]. In the present study, although there was no significant difference in overall mortality, we speculated that genetic factors did not decide the outcome of NAFLD. However, in subgroup analysis, PNPLA3 contributed to mortality in patients with non-diabetic or non-dyslipidemic complications. Therefore, we presumed that genetic influence would be greater in patients without lifestyle-related complications.
In contrast, patients with the ALDH2 AA genotype had lower activity of ALDH2 [14]. The protein level of ALDH2 is reportedly elevated in patients with nonalcoholic steatohepatitis [29]. Lower activity of ALDH2 SNP was related to mortality, particularly in patients without complications of dyslipidemia. ALDH2 contributes to alcohol metabolism and catalyzes the oxidation of aldehydes produced by lipid peroxidation, such as 4-hydroxy-2-nonenal. Thus, ALDH2 reduces the damage caused by reactive oxygen species and protects against oxidative stress. The reduced activity of ALDH2 likely hampers elimination of reactive oxygen species, promoting NAFLD development. In our previous study, patients without dyslipidemia and with hypertension had a poor prognosis [30]. We speculated that impaired liver function and advanced age may have played a role in some cases. Genetics may have also played a role in the outcome.
The gut microbiota contribute to the pathogenesis of nonalcoholic steatohepatitis; in particular, ethanol production by intestinal bacteria is increased in patients with nonalcoholic steatohepatitis [31]. Patients with nonalcoholic steatohepatitis were found to exhibit a significantly elevated blood ethanol level and enhanced hepatic alcohol metabolism [32]. Even among non-drinkers, patients with the ALDH2 non-GG genotype may have higher blood levels of ethanol and aldehyde production by intestinal bacteria, causing liver damage and extrahepatic malignancy.
Additionally, we speculate that a low level of alcohol consumption might increase cellular damage and cause mortality in patients with NAFLD. Regarding the cause of death, two patients with the ALDH2 non-GG genotype died of unknown causes, with suspected coronary disease. The activation of ALDH2 has been reported to attenuate atherosclerosis [33]. Furthermore, the non-GG genotype of ALDH2 appears to be associated with a higher frequency of coronary spasm [34]. However, we found that in patients with the complications of metabolic diseases, risk SNP carriers did not increase the mortality rate, and the ALDH2 non-GG genotype had a significantly poor prognosis among patients with hypertension. In contrast, modest alcohol consumption may be associated with reduced severity of myocardial infarction [35]; moreover, men who were moderate drinkers had a reduced risk of myocardial infarction [36]. However, patients with NAFLD who had regular alcohol intake, even in the safe range, had a higher risk of liver disease progression [37]. Therefore, even low alcohol intake is harmful to metabolically compromised patients. A genetic analysis should be conducted when assessing the safety of alcohol intake.
Fibrosis stage is the only histological feature that has been independently associated with liver-related events, including mortality and HCC [38–40]. Our results suggest that the serum level of albumin and genetic background was independently associated with mortality in patients with NAFLD. Lower serum albumin level indicates decreased albumin production by the liver and/or poor nutrition status and can contribute to the outcome of NAFLD patients. Furthermore, the risk SNPs of PNPLA3 and ALDH2, and in particular both risk SNPs were significantly associated with a poor prognosis.
This study had several limitations. First, it used a single-center observational design and involve a small number of events. Second, the effects of low levels of alcohol consumption (e.g., ‘social drinking’) and intestinal bacteria could not be evaluated. We did not evaluate the control of lifestyle-related diseases, duration of therapy, or changes in body weight. Further studies are needed to assess these factors as well as the effects of genetic background on mortality in Japanese patients with NAFLD.
5ConclusionsThe results suggested that genetic background affects the prognosis of NAFLD, particularly in the absence of complications of lifestyle-related diseases. ALDH2 SNP may be able to predict the outcome of NAFLD.AbbreviationsALDH2 aldehyde dehydrogenase 2 fibrosis-4 hepatocellular carcinoma non-alcoholic fatty liver disease patatin-like phospholipase 3 single nucleotide polymorphism
The datasets used and/or analyzed in this study are available from the corresponding author upon reasonable request.
Authors’ contributionsConception and design: TK and KT. Analysis and interpretation of the data: TK. Drafting of the manuscript: TK. Patient care, follow-up, and data acquisition: TK, TS, KK, MT, EH, and KT. All authors have read and approved the final manuscript. All authors agree to be accountable for all aspects of the study; questions related to the accuracy or integrity of any part of the study will be investigated and resolved appropriately.
Conflict of interestKT received research funding from Sumitomo Dainippon Pharma, Astellas Pharma, Eisai, Taiho Pharmaceutical, Chugai Pharmaceutical, Daiichi Sankyo Pharmaceutical, AbbVie GK, Takeda Pharmaceutical, Asahi Kasei Corporation, Ajinomoto, and Otsuka Pharmaceutical.
This work was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (#20K17035-0002) to T.S.



