




In patients with advanced liver disease with portal hypertension, portal-systemic collaterals contribute to circulatory disturbance, gastrointestinal hemorrhage, hepatic encephalopathy, ascites, hepatopulmonary syndrome and portopulmonary hypertension. Angiogenesis has a pivotal role in the formation of portal-systemic shunts. Recent research has defined many of the mediators and mechanisms involved in this angiogenic process, linking the central roles of hepatic stellate cells and endothelial cells. Studies of animal models have demonstrated the potential therapeutic impact of drugs to inhibit angiogenesis in cirrhosis. For example, inhibition of VEGF reduces portal pressure, hyperdynamic splanchnic circulation, portosystemic collateralization and liver fibrosis. An improved understanding of the role of other angiogenic factors provides hope for a novel targeted therapy for portal hypertension with a tolerable adverse effect profile.
Portal hypertension and the formation of portal-systemic collaterals are common features of advanced liver disease and give rise to many severe and life-threatening complications, including circulatory disturbance, gastrointestinal hemorrhage, hepatic encephalopathy and ascites. Up to 1% of adults in developed countries have cirrhosis,1,2 among whom the complications of portal hypertension are leading causes of death or liver transplantation. Gastroesophageal varices are present in up to 50% of adults with cirrhosis3 and if bleeding occurs, up to 20% of the initial bleeding episodes are fatal. Recurrent bleeding is common in the absence of secondary prophylactic therapy.4–6 Variceal hemorrhage also occurs commonly in children with chronic liver disease or portal vein obstruction.7–14 In children with biliary atresia, the incidence of variceal hemorrhage ranges from 17 to 29% over a five to 10 year period9,10 and is 50% in children who survive more than 10 years without liver transplantation.15
Prevention and improved management of portal hypertension and its complications are therefore important goals. In general, advances will be best achieved by future developments in two areas; firstly, the discovery and implementation of preventative and curative therapies for the underlying liver diseases that cause portal hypertension, and secondly the development of targeted therapies arising from an improved understanding of the mechanisms by which portal hypertension causes its complications.
Portal hypertension results from increased resistance to portal blood flow through the cirrhotic liver caused by the distortion of the liver architecture (secondary to fibrosis, nodule formation and vascular changes) and by alterations to hepatic sinusoidal cells and stellate cells that result in constriction of the hepatic sinusoids. Secondarily, a progressive splanchnic vasodilatation increases flow into the portal vein and further aggravates the portal hypertension.16
In the last decade, evidence has accumulated for the pivotal role of angiogenesis in the formation of portal-systemic shunts from pre-existing vasculature. Angiogenesis has also been linked with the progression of inflammation and fibrosis (Figure 1). In this review we will describe the knowledge gained from animal studies showing the importance of angiogenic mechanisms in the development of portal-systemic collaterals. We will summarize clinical studies of circulating concentrations of locally acting mediators of angiogenesis and their role in prediction of clinically relevant outcomes. Finally, we will suggest future areas for translational research to generate improvements in clinical care.
Key Players in Portal HypertensionSinusoidal and stellate cells
The liver parenchyma possesses two different types of microvascular structure. Firstly, branches of the large vessels such as the portal vein are lined by a continuous layer of endothelial cells lying on a basement membrane. Secondly, liver sinusoids are lined by fenestrated and discontinuous endothelial cells. When the sinusoidal lining cells face an injury, they lose their vasoprotective phenotype, becoming vasoconstrictor, proinflammatory and prothrombotic.17 The capillarization of the sinusoids in response to injury is associated with production of less nitric oxide and significantly more vasoconstrictor prostanoids.18,19
When NO availability is reduced, HSC become activated and develop a myofibroblast-like phenotype with a contractile response to vasoconstrictor molecules and diminished response to vasodilators.20,21 As cirrhosis develops, HSC proliferate and lay down extracellular matrix components, including collagen and proteoglycans. HSC are key players in portal hypertension and they express several angiogenic mediators, including VEGF, Ang-1, and their receptors VEGFR-1, VEGFR-2 and Tie-2.22 Thus changes in sinusoidal and stellate cells contribute to the increase in hepatic vascular resistance and the modulation of angiogenesis and fibrogenesis.
Hepatic fibrosisIn the liver, angiogenesis is postulated to contribute to portal hypertension by promoting fibrogenesis. Indeed, angiogenesis and fibrosis develop in parallel in a number of organ beds including the kidney and the lung.23 In the liver, neovasculature and overexpression of pro-angiogenic molecules have been detected in biopsies of patients with chronic viral infection, primary biliary cirrhosis and auto-immune hepatitis.24,25
Vascular structural changes within the liver are well established pathological hallmarks of chronic cirrhosis26,27 and may be reversible determinants of resistance and pressure regulation. In this context, studies in experimental models of cirrhosis have shown that liver fibrosis is decreased by treatment with inhibitors of angiogenic mediators such as VEGFR, PDGF and Ang-1.28,29 Moreover, in human liver samples, expression of angiogenic markers like Ang-1 and endothelial markers such as CD31 and von Willebrand factor correlate with the degree of hepatic fibrosis and presence of collagen 15A1.28,30 Similar findings were observed in animal studies using complementary models of liver fibrosis where fibrogenesis and angiogenesis develop in parallel during progression towards cirrhosis.29,31
Formation of Portal-Systemic CollateralsThe clinical manifestations of portal hypertension have a complex pathogenesis which includes splanchnic vasodilatation, increased portal venous inflow, increased hepatic resistance to portal venous flow, and the formation of portal-systemic collaterals.32,33 Collaterals contribute to the hemodynamic changes of portal hypertension that exacerbate fluid and salt retention and the formation of ascites; they allow the development of varices and the delivery of noxious substances from portal circulation directly to cerebral vasculature without hepatic detoxification, and thus contribute to hepatic encephalopathy;34 their presence may also contribute to the incompletely understood pathogenesis of hepatopulmonary syndrome and portopulmonary hypertension.35
The development of collaterals is associated with increased intraluminal pressure in the portal venous system. Esophageal varices only develop in cirrhosis once the hepatic venous pressure gradient (a measure of portal pressure in cirrhosis) is sustained above 10 mmHg.36 The mechanism by which increased portal pressure may result in the development of large collateral vessels has been the subject of recent research exploring the role of angiogenic mechanisms.
There are some key differences in the vasoactive pathways and mediators in the hepatic and splanchnic circulations. The increased vascular tone within the sinusoids of the cirrhotic liver is mediated by a limited quantity of vasodilators (such as NO) and an exaggerated amount of and response to vasoconstrictors. However, in the splanchnic vascular bed, there is an overproduction of vasodilators and a loss of response to vasoconstrictors. Endothelial cells in the splanchnic circulation generate an exaggerated amount of vasodilator messengers including nitric oxide, carbon monoxide, glucagon and prostacyclin.37–39 Splachnic hyperemia is further aggravated by the development of an extensive network of portal-systemic collateral venous vessels.
Mechanism of new blood vessel formationAngiogenesis is the formation of new blood vessels. It is an active process, dependent on growth factors, that takes place during growth and repair of injured tissues, but also in pathologic situations like tumour progression and in different inflammatory, fibro-proliferative and ischemic diseases.40
There are two main drivers of angiogenesis in all tissues: inflammation and hypoxia.41 During chronic tissue damage, the immune response leads to activation of endothelial cells, increased vascular permeability and production of chemokines that recruit more inflammatory cells.42–44 Local tissue hypoxia occurs due to altered blood flow in the setting of fibrosis and inflammation. Hypoxia activates angiogenesis through the actions of hypoxia-inducible factors (HIFs).45,46
Angiogenesis is mediated by complex interactions that can be divided into 4 steps:41
- •
Sprouting and budding of endothelial cells.
- •
Extracellular matrix degradation and endothelial cell migration.
- •
Endothelial cell proliferation, tube formation and branching and
- •
Vessel maintenance, maturation and stabilization.
Vascular collateral growth has been primarily studied in models of arterial occlusion, following which neighbouring small arteries may expand to enable restoration of blood supply to the potentially ischemic tissue supplied previously by the occluded artery.47 The extent of similarities between this process and the development of veno-venous, portal-systemic collaterals due to cirrhosis or portal vein thrombosis is unclear; there is little published data on the angiogenic mechanisms in venous collateral development.
Following arterial occlusion, blood pressure falls distal to the occlusion and a pressure gradient develops across pre-existing collateral vessels that establishes or increases blood flow in these collaterals. Blood flow creates shear stress, which triggers growth of arterial diameter until shear stress normalizes.48 The mechanisms linking shear stress to arterial growth are incompletely defined; endothelial cell activation occurs and transcription factors (nuclear factor κ-B, early growth response-1 and activator protein-1) and adhesion molecules (intracellular adhesion molecule-1 and vascular cell adhesion molecule-1) are upregulated.49,50 Proteolytic pathways are regulated in part by mechanical stress and are important in enabling collateral vessel growth through remodeling of the surrounding connective tissue.51 Production of monocyte chemoattractant protein-1 is increased by shear stress, and leads to the accumulation of monocytes around the collateral vessels.52,53 Vascular endothelial growth factor (VEGF) and placenta growth factor (PlGF) affect monocyte chemotaxis. VEGF also increases monocyte adhesion to endothelial cells and their subsequent transmigration.54,55 Furthermore, the renin-angiotensin-aldosterone system, insulin resistance and reactive oxygen species all contribute to the angiogenic process.56 Blockade of the renin-angiotensin-aldosterone system by an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker markedly attenuates liver fibrosis and hepatocellular carcinoma along with suppression of angiogenesis, insulin resistance and reactive oxygen species.56
Among the many mediators of the angiogenic process and its control, several are also of proven importance in liver organogenesis and regeneration, in liver fibrosis, or in other aspects of liver disease (Table 1). In the extensive literature describing the actions of these mediators, few studies focus on the growth of venous collaterals or the relevance of the mediators to the manifestations of portal hypertension. However, the importance of angiogenic mediators in the development of portal-systemic collaterals in portal hypertension has recently been identified.
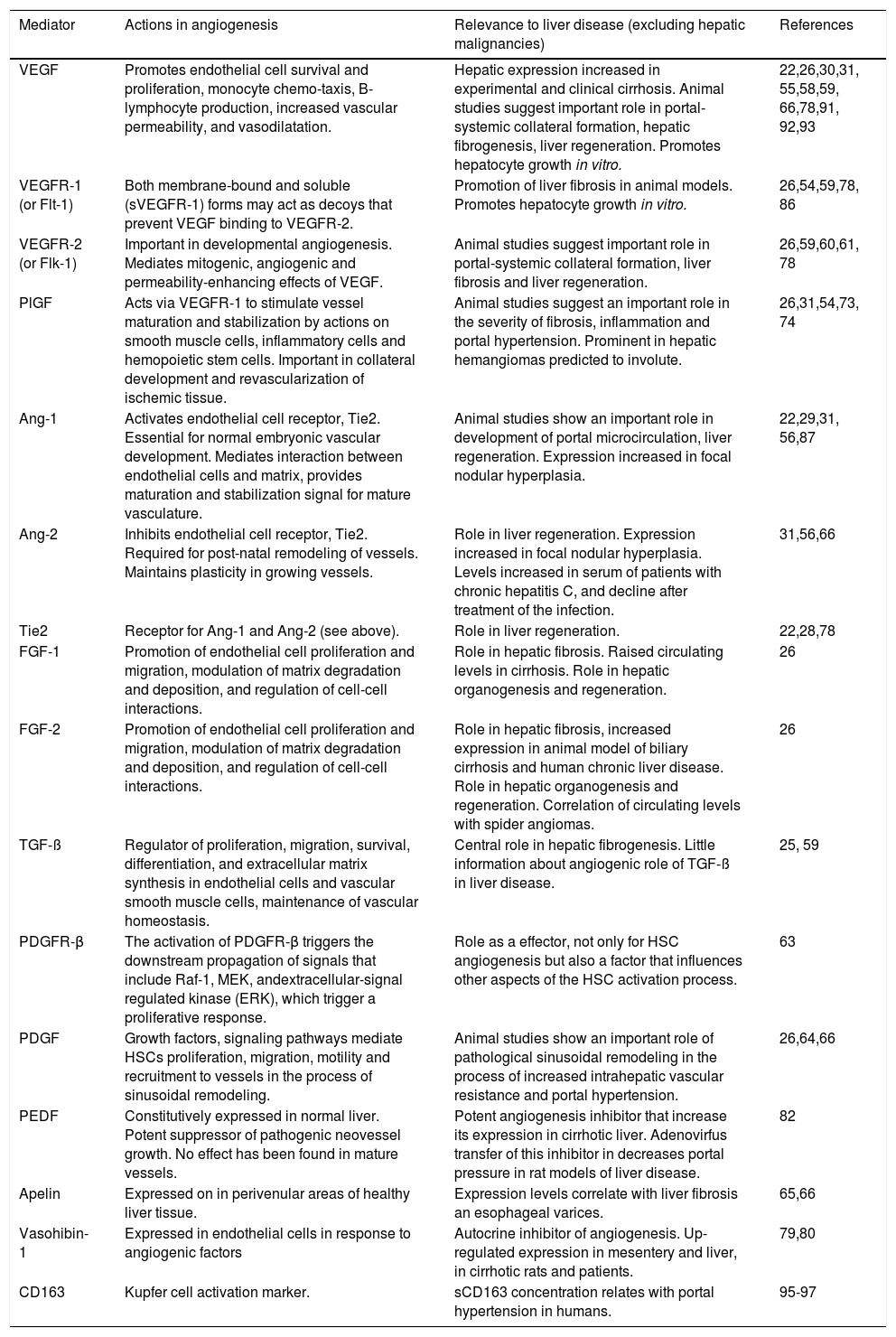
Mediators of angiogenesis and evidence for their role in liver disease.
| Mediator | Actions in angiogenesis | Relevance to liver disease (excluding hepatic malignancies) | References |
|---|---|---|---|
| VEGF | Promotes endothelial cell survival and proliferation, monocyte chemo-taxis, B-lymphocyte production, increased vascular permeability, and vasodilatation. | Hepatic expression increased in experimental and clinical cirrhosis. Animal studies suggest important role in portal-systemic collateral formation, hepatic fibrogenesis, liver regeneration. Promotes hepatocyte growth in vitro. | 22,26,30,31, 55,58,59, 66,78,91, 92,93 |
| VEGFR-1 (or Flt-1) | Both membrane-bound and soluble (sVEGFR-1) forms may act as decoys that prevent VEGF binding to VEGFR-2. | Promotion of liver fibrosis in animal models. Promotes hepatocyte growth in vitro. | 26,54,59,78, 86 |
| VEGFR-2 (or Flk-1) | Important in developmental angiogenesis. Mediates mitogenic, angiogenic and permeability-enhancing effects of VEGF. | Animal studies suggest important role in portal-systemic collateral formation, liver fibrosis and liver regeneration. | 26,59,60,61, 78 |
| PlGF | Acts via VEGFR-1 to stimulate vessel maturation and stabilization by actions on smooth muscle cells, inflammatory cells and hemopoietic stem cells. Important in collateral development and revascularization of ischemic tissue. | Animal studies suggest an important role in the severity of fibrosis, inflammation and portal hypertension. Prominent in hepatic hemangiomas predicted to involute. | 26,31,54,73, 74 |
| Ang-1 | Activates endothelial cell receptor, Tie2. Essential for normal embryonic vascular development. Mediates interaction between endothelial cells and matrix, provides maturation and stabilization signal for mature vasculature. | Animal studies show an important role in development of portal microcirculation, liver regeneration. Expression increased in focal nodular hyperplasia. | 22,29,31, 56,87 |
| Ang-2 | Inhibits endothelial cell receptor, Tie2. Required for post-natal remodeling of vessels. Maintains plasticity in growing vessels. | Role in liver regeneration. Expression increased in focal nodular hyperplasia. Levels increased in serum of patients with chronic hepatitis C, and decline after treatment of the infection. | 31,56,66 |
| Tie2 | Receptor for Ang-1 and Ang-2 (see above). | Role in liver regeneration. | 22,28,78 |
| FGF-1 | Promotion of endothelial cell proliferation and migration, modulation of matrix degradation and deposition, and regulation of cell-cell interactions. | Role in hepatic fibrosis. Raised circulating levels in cirrhosis. Role in hepatic organogenesis and regeneration. | 26 |
| FGF-2 | Promotion of endothelial cell proliferation and migration, modulation of matrix degradation and deposition, and regulation of cell-cell interactions. | Role in hepatic fibrosis, increased expression in animal model of biliary cirrhosis and human chronic liver disease. Role in hepatic organogenesis and regeneration. Correlation of circulating levels with spider angiomas. | 26 |
| TGF-ß | Regulator of proliferation, migration, survival, differentiation, and extracellular matrix synthesis in endothelial cells and vascular smooth muscle cells, maintenance of vascular homeostasis. | Central role in hepatic fibrogenesis. Little information about angiogenic role of TGF-ß in liver disease. | 25, 59 |
| PDGFR-β | The activation of PDGFR-β triggers the downstream propagation of signals that include Raf-1, MEK, andextracellular-signal regulated kinase (ERK), which trigger a proliferative response. | Role as a effector, not only for HSC angiogenesis but also a factor that influences other aspects of the HSC activation process. | 63 |
| PDGF | Growth factors, signaling pathways mediate HSCs proliferation, migration, motility and recruitment to vessels in the process of sinusoidal remodeling. | Animal studies show an important role of pathological sinusoidal remodeling in the process of increased intrahepatic vascular resistance and portal hypertension. | 26,64,66 |
| PEDF | Constitutively expressed in normal liver. Potent suppressor of pathogenic neovessel growth. No effect has been found in mature vessels. | Potent angiogenesis inhibitor that increase its expression in cirrhotic liver. Adenovirfus transfer of this inhibitor in decreases portal pressure in rat models of liver disease. | 82 |
| Apelin | Expressed on in perivenular areas of healthy liver tissue. | Expression levels correlate with liver fibrosis an esophageal varices. | 65,66 |
| Vasohibin-1 | Expressed in endothelial cells in response to angiogenic factors | Autocrine inhibitor of angiogenesis. Up-regulated expression in mesentery and liver, in cirrhotic rats and patients. | 79,80 |
| CD163 | Kupfer cell activation marker. | sCD163 concentration relates with portal hypertension in humans. | 95-97 |
Several animal studies suggest increased angiogenesis in portal hypertension,57,58 in which enlargement of portal-systemic collaterals occurs in response to angiogenic mediators. As occurs in other circumstances, the two principal drivers of angiogenesis in liver disease are inflammation and hypoxia.
Vascular endothelial growth factor (VEGF) promotes an extensive neovascularization in the mesenteric vascular bed in portal hypertension.26,59 In cirrhosis, the activated HSCs produce VEGF and express VEGF receptors on their surface, suggesting a paracrine and autocrine mode of action for this mediator.23
Fernandez, et al. demonstrated that VEGF-mediated angiogenesis causes formation of veno-venous, portal-systemic collaterals in portal hypertension through the receptor, VEGFR-2.60 Partial ligation of the portal vein of mice and rats produced a model of portal hypertension and enabled the study of the formation of portal-systemic collaterals, measured by splenic injection of radioactive microspheres and subsequent measurement of radioactivity in liver and lungs. The study tested the effects of a monoclonal antibody (DC101) against VEGFR-2 and an inhibitor of VEGF-2 function (SU5416) on portal-systemic collateral circulation and portal venous pressure. Intestinal and mesenteric VEGF expression increased approximately three-fold in portal hypertensive compared to sham-operated mice by day 7. VEGFR-2 and CD31 (an endothelial cell marker) expression were also significantly increased by partial portal vein ligation. Recurrent intraperitoneal injections of the monoclonal antibody against VEGFR-2 (DC101) resulted in 40-68% inhibition of collateral circulation over the ensuing days, but had no effect on portal pressure. CD31 (platelet endothelial cell adhesion molecule-1) expression in the splanchnic circulation was significantly reduced by DC101, suggesting reduced neovascularization in this location. Similar effects were noted when rats with partial portal vein ligation were given SU5416. Interestingly, portal pressure remained unchanged, probably due to the balanced effects of reduced portal inflow and relative increase in portal resistance, both due to the smaller collateral circulation.61
In experimental portal hypertension generated by partial portal vein-ligation, pharmacological inhibition of NAD(P)H oxidase reduced expression of VEGF and its receptor in the mesenteric circulation, in parallel with a decrease in the formation of portal systemic collaterals, a decrease in superior mesenteric arterial blood flow and increased resistance.62
Platelet derived growth factor (PDGF) and its receptor PDGFR-β are significantly up-regulated during portal hypertension in mesentery and cirrhotic liver and play an important role in the neovascularization.63 Endothelial cells produce PDGF and stimulate PDGFR-β on HSC, triggering their activation.64
Apelin is a widely expressed ligand of the G-proteincoupled APJ receptor that is known to influence angiogenesis. The apelin signaling pathway has been identified recently as an important contributor to liver dysfunction. Elevated mesenteric plasma levels of apelin in cirrhotic patients as well as expression of apelin and its receptor have been shown to occur in HSC located in the margin of the fibrous septa, in a murine model of portal hypertension.65 A recent study using an inhibitor of apelin demonstrated decrease in portal-systemic collateral formation as well as a reduced expression of several angiogenic mediators such as VEGF, PDGF and Ang-2 in the mesenteric circulation.66
Octreotide, a somatostatin analogue with potent antiangiogenic effect in models of experimental angiogenesis, was used to treat partial portal vein-ligated rats with portal hypertension. Although portal collateral formation was non-significantly reduced, splanchnic neovascularization and VEGF expression were decreased in a somatostatin receptor subtype 2 (SSTR2) dependent manner.67
The animal models used for these mechanistic studies of portal hypertension are summarized in table 2 and include those primarily involving parenchymal damage (e.g. carbon tetrachloride-induced hepatic toxicity) and those primarily increasing portal resistance (partial portal vein ligation, PPVL). These models mimic certain aspects of the specific human liver diseases associated with portal hypertension, but are not ideal models to provide an accurate reflection of portal hypertension in human disease. For example, the PPVL models, whilst nicely mimicking splanchnic venous hypertension, may not provide the local and systemic milieu of inflammatory and other mediators that comprise the cirrhotic state in humans with liver disease. Similarly, a carbon tetrachloride hepatotoxicity model may not accurately reflect the intrahepatic and splanchnic environment associated with cirrhotic hepatitis C or cholestatic liver disease. Animal study results must therefore be interpreted with these limitations in mind, including those exploring the effects of therapies that target angiogenesis in portal hypertension.
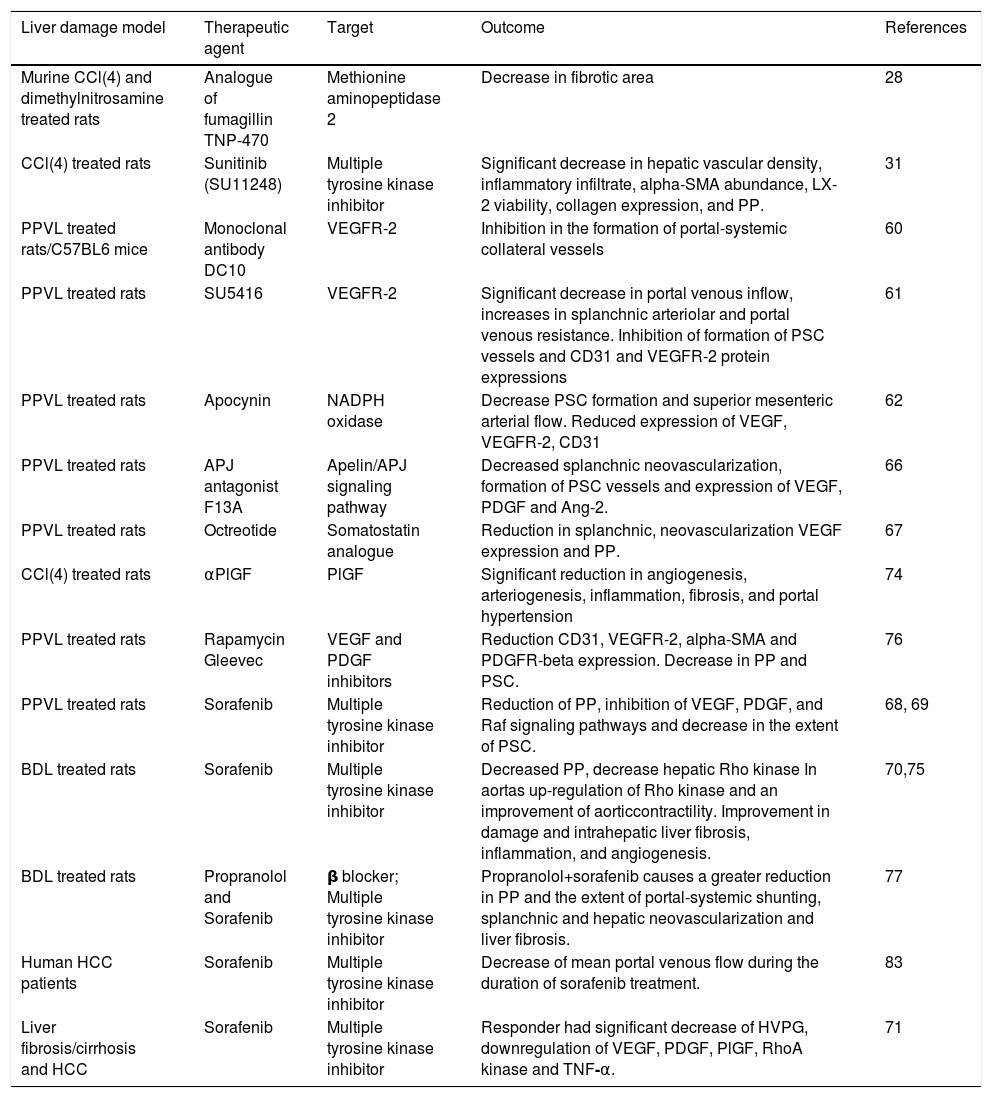
Portal hypertension interventional models summarized in this study.
| Liver damage model | Therapeutic agent | Target | Outcome | References |
|---|---|---|---|---|
| Murine CCl(4) and dimethylnitrosamine treated rats | Analogue of fumagillin TNP-470 | Methionine aminopeptidase 2 | Decrease in fibrotic area | 28 |
| CCl(4) treated rats | Sunitinib (SU11248) | Multiple tyrosine kinase inhibitor | Significant decrease in hepatic vascular density, inflammatory infiltrate, alpha-SMA abundance, LX-2 viability, collagen expression, and PP. | 31 |
| PPVL treated rats/C57BL6 mice | Monoclonal antibody DC10 | VEGFR-2 | Inhibition in the formation of portal-systemic collateral vessels | 60 |
| PPVL treated rats | SU5416 | VEGFR-2 | Significant decrease in portal venous inflow, increases in splanchnic arteriolar and portal venous resistance. Inhibition of formation of PSC vessels and CD31 and VEGFR-2 protein expressions | 61 |
| PPVL treated rats | Apocynin | NADPH oxidase | Decrease PSC formation and superior mesenteric arterial flow. Reduced expression of VEGF, VEGFR-2, CD31 | 62 |
| PPVL treated rats | APJ antagonist F13A | Apelin/APJ signaling pathway | Decreased splanchnic neovascularization, formation of PSC vessels and expression of VEGF, PDGF and Ang-2. | 66 |
| PPVL treated rats | Octreotide | Somatostatin analogue | Reduction in splanchnic, neovascularization VEGF expression and PP. | 67 |
| CCl(4) treated rats | αPlGF | PlGF | Significant reduction in angiogenesis, arteriogenesis, inflammation, fibrosis, and portal hypertension | 74 |
| PPVL treated rats | Rapamycin Gleevec | VEGF and PDGF inhibitors | Reduction CD31, VEGFR-2, alpha-SMA and PDGFR-beta expression. Decrease in PP and PSC. | 76 |
| PPVL treated rats | Sorafenib | Multiple tyrosine kinase inhibitor | Reduction of PP, inhibition of VEGF, PDGF, and Raf signaling pathways and decrease in the extent of PSC. | 68, 69 |
| BDL treated rats | Sorafenib | Multiple tyrosine kinase inhibitor | Decreased PP, decrease hepatic Rho kinase In aortas up-regulation of Rho kinase and an improvement of aorticcontractility. Improvement in damage and intrahepatic liver fibrosis, inflammation, and angiogenesis. | 70,75 |
| BDL treated rats | Propranolol and Sorafenib | β blocker; Multiple tyrosine kinase inhibitor | Propranolol+sorafenib causes a greater reduction in PP and the extent of portal-systemic shunting, splanchnic and hepatic neovascularization and liver fibrosis. | 77 |
| Human HCC patients | Sorafenib | Multiple tyrosine kinase inhibitor | Decrease of mean portal venous flow during the duration of sorafenib treatment. | 83 |
| Liver fibrosis/cirrhosis and HCC | Sorafenib | Multiple tyrosine kinase inhibitor | Responder had significant decrease of HVPG, downregulation of VEGF, PDGF, PlGF, RhoA kinase and TNF-α. | 71 |
*CCL: carbon chloride. PPVL: partial portal vein ligation. BDL: bile duct ligation. SMA: smooth muscle actin. PP: portal pressure. VEGF: vascular endothelial growth factor. PSC: portosystemic collaterals. PDGF: platelet-derived growth factor. ANG: angiopoietin. PlGF: placental growth factor. HCC: hepatocellular carcinoma. HVPG: hepatic venous pressure gradient. TNF: tumor necrosis factor.
A potential role for therapies that target angiogenesis is suggested by its central role in the development of portal hypertension and its complications. However, a successful therapy of this sort would need to avoid adverse effects due to inhibition of the normal physiological angiogenesis process, for example during vessel growth in childhood or during the repair of injured tissues (Table 2).
Inhibition of VEGF by several drugs has been shown to be effective in reducing portal pressure, reducing the hyperdynamic splanchnic circulation, reducing portosystemic collateralization60,68 and improving liver fibrosis.31,69,70 Potentially concerning adverse effects have included endothelial injury and enhanced bleeding risk.71,72 A more selective approach to blocking angiogenesis in cirrhosis may be required.
Placental growth factor (PlGF) forms an interesting target because its role is restricted to pathological conditions, suggesting that fewer adverse effects may be expected. PlGF antagonists have shown promising results in preventing angiogenesis, inflammation, portal hypertension and fibrosis in an animal model.73,74
Simultaneous therapies impacting multiple targets in both angiogenesis and inflammation have been shown to be beneficial in inhibiting the progression of fibrosis to cirrhosis in animal models. The validity of this approach was demonstrated in cirrhotic rats in which sunitinib and sorafenib, two inhibitors of tyrosine kinase receptors (RTKs) that target the platelet-derived growth factor (PDGF) and VEGF signaling pathways, produced a reduction in the degree of hepatic angiogenesis, fibrosis, and inflammation, as well as a significant decrease in portal pressure.31,69,75,76 These specific agents may also inhibit PDGFR-β, which effects not only angiogenesis but also HSC activation. Treatment combining the use of sorafenib with propranolol markedly reduced the extent of portal-systemic shunting, splanchnic and hepatic neovascularization, and liver fibrosis in comparison to any of the individual treatments.77 Drugs that specifically inhibit angiogenesis by targeting molecules not apparently directly involved in the fibrogenic pathway, like VEGFR-2 or Tie2, also induce a decrease in hepatic fibrosis,29,78 providing further evidence for the importance of angiogenesis in the fibrogenic process.
Finally, promising early results have been obtained in studies that aim to augment the endogenous inhibitors of angiogenesis, such as vasohibin-1 and pigment epithelium derived factor.79–82
In humans, the multiple tyrosine kinase inhibitor sorafenib has been approved for use in treatment of hepatocellular carcinoma (HCC). Scarce data is available about the effect of this inhibitor in portal pressure or liver fibrosis. Two different studies analysed the effect of sorafenib in 7/13 patients with HCC with emphasis on hepatic venous pressure gradient, porto-collateral circulation and fibrotic and angiogenic factor expression. The first study showed a reduced portal venous flow during the duration of sorafenib treatment that was reversible upon sorafenib removal.83 In the second study 36% of portal hypertensive patients showed a response to the treatment characterized by a significant decrease of HVPG. A down regulation of intrahepatic VEGF, PDGF, PlGF, RhoA kinase and TNFα mRNA expression was observed only in the responder subgroups of patients.71 Both studies were performed in a HCC setting with very small sample sizes and no control groups so additional information is required to determine the potential positive effects of this treatment in portal hypertensive patients.
Circulating Concentrations of Angiogenic Mediators: Biomarkers of Disease SeverityMediators involved in the control of angiogenesis act in the local mesenteric and hepatic environments to attract and activate cells and to change the surrounding connective tissue. Although their actions are predominantly autocrine and paracrine, production of angiogenic mediators by endothelial and neighbouring inflammatory cells may lead to significant concentrations detectable in blood. Circulating angiogenic mediators that may act as biomarkers for the severity of portal hypertension and its complications would be of great clinical value. For example, in patients with hepatocellular carcinoma and other cancers, the concentrations of circulating angiogenic factors predict clinical outcomes, including tumour progression and patient survival.84–89
There is conflicting evidence as to what extent concentrations of VEGF in peripheral blood correlate with the severity of chronic liver disease or portal hypertension.90–93 Studies of circulating VEGF concentrations in adults with liver disease have been confounded by measurement of VEGF in serum, where its concentration is affected by platelet count and function, both of which may be abnormal in portal hypertension; VEGF concentration in plasma is more independent from platelet count.91–93 However, there may be differences in regional VEGF production in portal hypertension, as has been found with nitric oxide.33 Therefore, the importance of circulating angiogenic mediators in the pathophysiology of portal hypertension may be better characterized by studying their concentrations in portal blood.94–96
ConclusionsThe formation of portal-systemic collateral vessels contributes to the clinical complications of portal hypertension and causes significant morbidity and mortality. Current therapeutic approaches do not prevent the formation of collaterals, but aim to reduce their serious clinical impact. The growing understanding of the importance of angiogenesis in portal hypertension is identifying potential novel therapeutic targets. Results of further studies of antiangiogenic agents in humans with cirrhosis are eagerly awaited.
SupportedThis work was supported by Fondecyt grant #11121146.



