










Relationship between cirrhosis and renal dysfunction is not yet fully understood. A model of cirrhosis with acute hepatic and renal damage (RF), produced by CCl4 in rats, with hemodynamic and renal functional alterations, similar to those observed in decompensated cirrhosis (DC) in man, was used to study chemical nephrotoxicity in animals. We performed in male Wistar rats hepatic and renal functional and hemodynamic studies in control, cirrhotic and decompensated cirrhotic (DC) groups. Cirrhosis was induced with carbon tetrachloride by chronic administration. Association between liver and renal functional alterations was detected in rats with decompensated cirrhosis, showing fall in mean arterial pressure and reduction of glomerular filtration rate and filtration fraction. Renal hemodynamics did not change in cirrhotic rats, similarly to what occurs in compensated cirrhotic patients. However, DC rats exhibited increased sodium, glucose and phosphate urinary excretions and decreased ATP in renal cortex. DC animals had severe hypoglycemia. There was an extensive liver fibrosis. Glomeruli had hypercellularity and tubules showed extensive vacuolization in cirrhotic and DC rats. The present study suggests that in this model, damage typical of acute tubular necrosis ensues in cirrhotic rats. We describe functional and morphological damage in liver and kidney in a model of cirrhosis that might predispose to the development of acute renal failure when an individual with hepatic damage is exposed in acute way to chemical toxicants.
Alterations in kidney structure and function are frequently found in severe liver disease and once liver function falls below a critical threshold, sodium retention occurs followed by ascites, associated with profound disturbances of splachnic and systemic hemodynamics which in turn may affect renal function.1,2 As disease progresses, constriction of intrarenal vascular system favors marked sodium and water retention, leading to refractory ascites, a progressive rise in plasma creatinine levels and reduction of renal clearances (decompensated cirrhosis). Persistent renal hypoxia may also induce tubular damage.3
Development of renal failure in patients with liver failure is frequent, it occurs in approximately the 55% of patients. This complication may result when the cirrhotic individual is exposed to xenobiotics, either therapeutic drugs or environmental pollutants, especially heavy metals. Renal function is rarely restored in the absence of hepatic recovery.4 Administration of carbon tetrachloride (CCl4) to rats results in a reproducible experimental model of cirrhosis, that resembles the disease in humans and provides a tool to study liver-kidney interrelationships.5,6Previously we have performed an experimental model of acute liver and renal damage, produced by intragastric administration of a single dose of CCl4 to cirrhotic rats. In this experimental model there are hemodynamic and renal functional alterations similar to those observed in the human with decompensated cirrhosis. This model is useful to study the pathogenesis of renal failure associated with liver damage when the hepatic function decrease after an acute liver damage.7
In our present work, we used our model to further characterize the systemic and renal hemodynamics, histopathological and functional changes of proximal tubular cells (renal sodium handling, phosphate and glucose excretion rates) as well as the function of cells from the distal nephron (renal water handling), in cirrhotic and in cirrhotic rats with acute liver damage induced by carbon tetrachloride.
Material and methodsAnimals:Male Wistar rats were maintained under 12 h day/ night light cycles fed ad libitum with rodent lab diet (Ralston Rations-Kansas, KA). Animals were housed in the animal facility of University Autonomous of Aguascalientes and all animal studies were conducted in accordance with the principles and procedures outlined in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Induction of liver cirrhosis: Liver cirrhosis was achieved in 100 male Wistar rats (40-60 g initial weight; 250 – 350 g final weight) by intraperitoneal injection of 0.2 ml of a mixture of CCl4 (J.T. Baker USA) and mineral oil (Sigma Chemical Company, St. Louis MO) 3 times per week during 8 weeks, progressively increasing CCl4 concentration until the fourth week of treatment.7 The percentages of CCl4 in mineral oil (v/v) were as follows: week 1, 13%; week 2, 16%; week 3, 20% and weeks 4 to 8, 25%. Age and sex matched control animals received the same volume of mineral oil.7
Induction of acute renal damage: At the end of eight weeks of chronic CCl4 treatment and after 6 days of the last chronic CCl4 dose, cirrhotic rats received a single intragastric dose of a 1:1 (v/v) mixture of CCl4 /corn oil (0.5 mL per 100 g body weight). This acute treatment change cirrhotic by decompensated cirrosis rats and induce acute renal failure in the animals.7-9
Liver function tests: Liver damage was assessed by measurements of: a) serum albumin,10,11 b) aspartate ami-notransferase (AST) and alanine aminotransferase (ALT) activities12 and c) total proteins. The method of Lowry et al. modified by Peterson13 was used for protein determinations using bovine albumin as standard.
Renal function tests: Animals were divided into three groups: control, cirrhotic and cirrhotic plus acute renal damage (decompensated cirrhosis). In those groups, we studied: (A) glomerular filtration rate (GFR) and plasma renal flow (PRF); (B) renal sodium handling (C) renal cortical Na+-K+- ATPase activity and tissue ATP concentrations, (D) renal glucose and phosphate excretion rates and (E) renal water handling.
Histological samples examination: Four animals per group (control and CCl4-treated) were anesthetized with sodium pentobarbital and intravascularly perfused with phosphate buffer solution (PBS), pH 7.4, containing heparine (1,000 UI/L) and procaine (1 g/L) for rinsing of livers and kidneys. For in situ fixation, tissue was perfused with PBS, pH 7.4, containing 10% formaldehyde (v/v). Livers and kidneys were removed and fixed for 24 h in the same fixing solution. 5-μm sections were stained with hematoxylin and eosin. Tissue sections were evaluated by optical microscopy. Examiner was unaware of the group to which samples belong.
Renal hemodynamic and sodium handling assays. Glomerular filtration rate (GFR) and renal plasma flow (RPF) were estimated by inulin and p-aminohippurate clearances, as previously described.14 Arterial blood pressure (BP) was measured with a Statham P-23A transducer and recorded in a RP Beckman Dynograph. Sodium, inulin and PAH were measured by flame photometry, anthrone method and spectrophotometry, as previously described.14,15
Renal cortex ATPase activities and tissue ATP concentrations: Renal cortex was separated from medulla and homogenized in solution containing (mM): 240 sucrose, 1 EDTA and 10 Tris-HCl, pH 7.5 (20 % w/v homogenate). ATPase activities were measured according to Quigley and Gotterer 16 ATP concentration was determined from renal cortex, according to the method of Bucher, modified by Adams.
Phosphate and glucose excretion: Cirrhotic, DC and age-matched control rats were kept in individual metabolic cages. Twenty four hour urine samples were collected and urinary volume was recorded. Urinary and plasma phosphate and glucose were measured by Sumner method adapted for microsamples and by the Trinder method, respectively. The urinary/plasma phosphate and the urinary/plasma glucose ratios were calculated.
Renal water handling: Water handling was estimated by osmolal and free-water clearances. Urine samples were collected and total urinary volume of each rat was recorded. Plasma and urine osmolalities were measured by cryoscopy (OSMETTE, Model 5004). Osmolal (Cosm) and free-water (CH2O) clearances were calculated as follows:
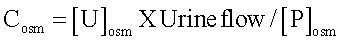
where [U]osm and [P]osm are urine and plasmatic-osmolalities, respectively, and urine flow is expressed in mL/ min. Free-water clearance was calculated as follows:
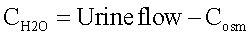
Statistical analyses: Results show means ± SD. Data were analyzed using ANOVA followed by Tukey Kramer test. Differences were considered significant when p < 0. 05.
ResultsInduction of cirrhosis resulted in approximately 30% mortality and when renal failure (RF) ensued, mortality additionally increased to 60%. Ascites was currently present.
Morphological studiesMorphology was normal in livers and kidneys of control animals (healthy animals) (Figures 1aand1d). In contrast, cirrhotic and decompensated rats with renal failure (RF) showed extensive liver fibrosis confirming that hepatic cirrhosis was attained (Figures 1band1c). Kidneys of cirrhotic and DC with RF-rats showed degenerative changes (Figures 1eand1f). Glomeruli were denser in both cirrhotic and DC with RF-rats, than in control group. In the cirrhotic group, there was extensive mesangial hypercellularity and podocytes showed hyperchromatic nuclei. Tubules exhibited fine and extensive vacuolization. In DC with RF-group, there was severe glomerular damage with increased mesangial space and hyperchromatic nuclei of podocytes; tubules showed cloudy swelling and degeneration (Figure 1f).
 Control group. Hepatocyte (arrows), kupffer cells (arrowheads), and sinusoidal space (asterisk) disclose normal liver organization and structure. (b) Liver exhibited extensive damage with necrosis and fibrosis (arrows), hyperplasia of kupffer cells is observed (peak arrow) and most of liver morphology was altered by cirrhosis. (c) Extensive and diffuse damage, with vesicular degeneration and necrosis (arrows) was observed in decompensated cirrhosis with renal failure group (dc+rf). (d) No morphological abnormalities were observed in the kidney of control animals: glomerular area (asterisk), mesangial space (small arrow), podocyte (arrowhead), proximal and distal tubules (arrows). (e) Cirrhotic animals exhibited hyperchromatic podocyte (arrowhead) and large mesangial spaces (small arrow); cloudy swelling (arrows) and hydropic degeneration (double arrow) were observed in the tubules. (f) In dc+rf group extensive mesangial hypercellularity with hyperchromatic nuclei (small and arrowhead), severe damage with structural modification in all the glomerular area were observed. tubules exhibited loss of brush border, cloudy swelling and cellular fragments inside the lumen (arrows).")
Histopathologic evaluation of rat liver and kidney. (a) Control group. Hepatocyte (arrows), kupffer cells (arrowheads), and sinusoidal space (asterisk) disclose normal liver organization and structure. (b) Liver exhibited extensive damage with necrosis and fibrosis (arrows), hyperplasia of kupffer cells is observed (peak arrow) and most of liver morphology was altered by cirrhosis. (c) Extensive and diffuse damage, with vesicular degeneration and necrosis (arrows) was observed in decompensated cirrhosis with renal failure group (dc+rf). (d) No morphological abnormalities were observed in the kidney of control animals: glomerular area (asterisk), mesangial space (small arrow), podocyte (arrowhead), proximal and distal tubules (arrows). (e) Cirrhotic animals exhibited hyperchromatic podocyte (arrowhead) and large mesangial spaces (small arrow); cloudy swelling (arrows) and hydropic degeneration (double arrow) were observed in the tubules. (f) In dc+rf group extensive mesangial hypercellularity with hyperchromatic nuclei (small and arrowhead), severe damage with structural modification in all the glomerular area were observed. tubules exhibited loss of brush border, cloudy swelling and cellular fragments inside the lumen (arrows).
As expected, in cirrhotic and DC with RF rats, serum albumin concentrations were lower (33.6% and 60%, respectively) than in controls. In DC with RF group serum albumin further decreased 39.8 %, in relation to cirrhotic rats (Figure 2, panel A). Total protein decreased 50 % and 33.6 % in the cirrhotic and DC with RF rats, respectively, as compared to control values (Figure 2, panel B). AST and ALT in DC with RF group and ALT in cirrhotic rats increased significantly, compared to controls, and in the DC with RF group AST and ALT serum activities further increased 255 % and 238 %, as compared with cirrhotic rats (Figure 2, panel C and D).
, produced by CCl4. Panel A) Serum albumin in control (empty bar), cirrhotic (hatched bar) and rats with acute hepatic and renal damage (fill bar).Panel B) Total plasma protein content, symbols as in panel A: C): Aspartate aminotransferase in the same groups of rats previously described; D): Alanine aminotransferase, ALT. Mean ± SEM are shown, *p < 0.05, **p < 0.01 and ***p < 0.001, when compared with control B) group.")
Hepatic function studies in control, cirrhotic and decompensated cirrhotic rats with renal failure (RF), produced by CCl4. Panel A) Serum albumin in control (empty bar), cirrhotic (hatched bar) and rats with acute hepatic and renal damage (fill bar).Panel B) Total plasma protein content, symbols as in panel A: C): Aspartate aminotransferase in the same groups of rats previously described; D): Alanine aminotransferase, ALT. Mean ± SEM are shown, *p < 0.05, **p < 0.01 and ***p < 0.001, when compared with control B) group.
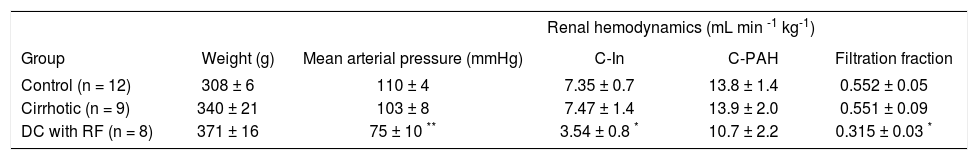
Mean arterial blood pressure (MAP) was similar in control and cirrhotic rats. By contrast, we also observed a fall in the MAP in the DC rats with RF, suggesting reduction in renal perfusion pressure (Table I). GFR also fell by 52 % in DC with RF group, as compared to control and cirrhotic animals, while no differences were observed between these two groups. Moreover, filtration fraction fell by 43% in group of DC with RF and did not change in cirrhotic rats (Table I). Renal plasma flow (RPF) did not show differences.
Mean arterial blood pressure and renal hemodynamic data from control, cirrhotic and decompensed cirrhotic rats with renal failure, produced by CCl4.
| Renal hemodynamics (mL min -1 kg-1) | |||||
|---|---|---|---|---|---|
| Group | Weight (g) | Mean arterial pressure (mmHg) | C-In | C-PAH | Filtration fraction |
| Control (n = 12) | 308 ± 6 | 110 ± 4 | 7.35 ± 0.7 | 13.8 ± 1.4 | 0.552 ± 0.05 |
| Cirrhotic (n = 9) | 340 ± 21 | 103 ± 8 | 7.47 ± 1.4 | 13.9 ± 2.0 | 0.551 ± 0.09 |
| DC with RF (n = 8) | 371 ± 16 | 75 ± 10 ** | 3.54 ± 0.8 * | 10.7 ± 2.2 | 0.315 ± 0.03 * |
Probably related to decrement in MAP, filtered sodium load decreased in DC rats with RF (756 ± 184 μEq/min/kg) as compared to control and cirrhotic rats (1032 ± 94 and 1117 ± 214 μEq/min/kg, respectively). In spite of diminished filtered sodium load, there was an increment in the fractional excretion of sodium (FeNa), in DC rats with RF. FeNa increased by 135% in DC rats with RF as compared to control rats and, excretion was 68% higher when compared to the cirrhotic group (Figure 3), indicating severe alteration of the tubular handling of sodium.
, produced by CCl4. Hepatic cirrhosis was produced in Wistar male rats with CCl4 (ip, for 8 weeks) and acute hepatic and renal damage was induced by an additional dose of CCl4 given orally to cirrhotic rats. Mean ± SEM are shown, **p < 0.01, when compared with control group.")
Sodium renal handling in control, cirrhotic and decompensated cirrhotic rats with renal damage (RF), produced by CCl4. Hepatic cirrhosis was produced in Wistar male rats with CCl4 (ip, for 8 weeks) and acute hepatic and renal damage was induced by an additional dose of CCl4 given orally to cirrhotic rats. Mean ± SEM are shown, **p < 0.01, when compared with control group.
Renal cortical ATP concentrations decreased in both CCl4-treated groups. We also observed fall of ATP significantly, in tissue of cirrhotic DC rats with RF, 62% and 71%, respectively (Figure 4). By contrast, both groups, cirrhotic and DC rats with RF, showed renal cortical ATPases (Total, Mg++-dependent and Na+-K+-dependent) that were significantly unaffected. Na+-K+-ATPase activity changed from a control value of 23 ± 3.3 to 32.5 ± 4.6 and 32.7 ± 10, nmol Pi min-1 mg-1, in cirrhotic and DC rats with RF respectively. These findings suggest that low tissue concentration of ATP was probably related to nucleotide altered synthesis.
, produced by CCl4. Symbols as in figure 3. Mean ± SEM of the percent change are shown, ***p < 0.001, when compared with control group.")
Renal cortical ATP from control, cirrhotic and decompensated cirrhotic rats with renal failure (RF), produced by CCl4. Symbols as in figure 3. Mean ± SEM of the percent change are shown, ***p < 0.001, when compared with control group.
However, glucose urine/plasma ratio and phosphate urine/plasma ratio were markedly higher in both groups, cirrhotic and DC rats with RF, as compared to control values, indicating marked urinary losses of glucose and phosphate. Indeed, urine/plasma glucose ratio increased 7.5fold in cirrhotic rats and 21.5-fold in DC rats with RF. Also, there was a significant difference in the urine/plasma glucose ratio values between DC rats and cirrhotic rats with RF (p < 0.001) (Table II). Related to phosphate urine/ plasma ratio, it increased 1.4-fold in cirrhotic rats (not significant) and 5.7-fold in DC rats group with RF (Table II). On the other hand, the glucose urinary concentration increased significantly in both cirrhotic (522 %) and RF rats (855 %) (Figure 5B). Because of these changes, serum glucose concentration markedly decreased in DC rats with RF group (54 %), without significant differences between control and cirrhotic rats (Figure 5A).
The urine/plasma ratio of glucose and phosphates from control, cirrhotic and decompensated cirrhotic rats with renal failure, produced by CCl4.
| Group | Urine/plasma glucose ratio | Urine/plasma phosphate ratio |
| Control (n = 10) | 0.026 ± 0.004 | 3.74 ± 0.34 |
| Cirrhotic (n = 10) | 0.196 ± 0.030 * | 5.13 ± 0.45 |
| DC with RF (n = 10) | 0.561 ± 0.075 ** | 21.18 ± 2.13 ** |
, produced by CCl4. Panel A) Serum glucose was measured at the end of 8 weeks treatment in control and cirrhotic rats, and at week 9, after acute hepatic and renal failure (RF) ensued. Panel B) Urine glucose was measured in the same time and groups before mentioned. Mean ± SEM are shown, ** p < 0.01 and *** p < 0.001, when compared with control group.")
Serum and urine glucose concentrations from control, cirrhotic and decompensated cirrhotic rats with renal failure (RF), produced by CCl4. Panel A) Serum glucose was measured at the end of 8 weeks treatment in control and cirrhotic rats, and at week 9, after acute hepatic and renal failure (RF) ensued. Panel B) Urine glucose was measured in the same time and groups before mentioned. Mean ± SEM are shown, ** p < 0.01 and *** p < 0.001, when compared with control group.
Significant decrease in urinary flow was observed in both groups cirrhotic and DC rats with RF (3.8 ± 0.35 and 3.5 ± 0.32 μL/min, respectively) as compared to control group (16.3 ± 1.07 μL/min). Urine osmolalities were higher in cirrhotic (1597 ± 170) and DC rats with RF (2290 ± 138), than in control group (595 ± 17 mOsmol/kg). Moreover, osmolal clearances were lower in cirrhotic and DC rats with RF (20 ± 1.5 and 26 ± 1.6 μL/min, respectively) than in control group (33 ± 1.6 μL/min). Free-water clearance decreased significantly in DC rats with RF when compared to control and cirrhotic groups (Figure 6).
Discussion, produced by CCl4. Free-water clearances (CH20) were estimated in the three experimental groups previously described. Mean ± SEM are shown, *p < 0.05.")
Hemodynamic and functional renal alterations in DC rats with RF were more severe than in cirrhotic rats. When additional acute liver damage was induced in cirrhotic rats, intense hemodynamic and renal functional alterations ensued. We observed fall in mean arterial pressure (MAP) probably due to diminished peripheral vascular resistance and significant reduction of GFR and filtration fraction in DC rats with RF, in a similar fashion as reported to cirrhotic rats 17. In contrast, renal hemodynamics did not change in cirrhotic rats, similarly to what occurs in compensated cirrhosis in humans.
Renal response to decreased blood pressure is altered in severe liver disease with activation of sympathetic system and increased synthesis of several renal vasoconstrictor compounds (endothelin, thromboxane A2 and F2-iso-prostanes). Activated sympathetic nervous system, and presumably other renal vasoconstricting agents, cause a rightward shift in the autoregulatory curve, making renal blood flow more pressure-dependent. Thus, even modest decreases in MAP may result in a marked fall of renal blood flow.4 Hemodynamic alterations led to the use of splanchnic vasoconstrictors, such as terlipressin.18 It has been reported that GFR and RPF are unchanged in cirrhotic rats.19 These findings are in agreement with our renal hemodynamic data in cirrhotic and control rats.
We observed increased excretions of sodium, glucose and phosphate in DC rats with RF. For glucose, the consequence was glycosuria, with marked hypoglycemia in DC rats with RF. Moreover, both glucose urine/plasma ratio and phosphate urine/plasma ratio were also higher in DC rats with RF than in cirrhotic rats. Increased FeNa, intense glycosuria (with severe hypoglycemia) and phosphaturia indicate severe functional alteration in proximal segments of the nephron. Morphological alterations in proximal tubules were in accordance with these functional alterations.
ATP tissue content was significantly reduced in DC rats with RF, reflecting scarcity of the main energetic source for sodium, glucose and phosphate transport. These severe metabolic alterations might partially explain the high mortality observed in the RF-experimental model. Relevance of our findings in human RF remains to be defined.
Proximal reabsorption of filtered sodium occurs in two steps: sodium enters the cell across the apical membrane via transmembrane carriers (SGLT 1 and 2) that also reabsorb glucose, coupled to the transport of phosphate or aminoacids, by selective sodium channels or through the paracellular pathway. Sodium is then returned to systemic circulation by the (Na+-K+)- ATPase located at the basolateral membrane.20,21 Glucose and phosphate reabsorption are coupled to sodium transport, via transmembrane co-transporters.22,23 Therefore increased urinary losses of sodium, glucose and phosphate are indicative of severe proximal tubular damage.24
Renal retention of sodium is considered to be responsible for edema and ascites in hepatic cirrhosis.25 The mechanisms of abnormal tubular sodium reabsorption in cirrhosis are multifactorial. Overactivity of the renin-angiotensin-aldosterone system and the sympathetic nervous system might play a role in this abnormality.26 Increased urinary sodium excretion has also been reported in cirrhosis. Approximately 10% to 20% cirrhotic patients with ascites spontaneously eliminate relatively large amounts of sodium in the urine.25 Decrease in tubular reabsorption is generally regarded as evidence of intrinsic tubular dysfunction. To explain irreversibility of DC damage with RF, it has been also suggested that this illness may be complicated by acute tubular necrosis.3 Histological alterations in the kidney of DC patients with RF, similar to those observed during acute tubular necrosis (ATN), with tubular degeneration and interstitial leukocyte infiltration have been reported.27,28 We observed degenerative changes in cirrhotic and DC rats with RF (Figure 1).
Moreover, depletion of ATP may increase cytosolic calcium, which activates proteases and phospholipase A2 with increased synthesis of leukotrienes that might induce renal ischemia.3 In accordance, increased production of cysteinyl leukotrienes in RF has been reported.29 Cellular energy depletion results in rapid loss of polarity in proximal tubule cells. (Na+-K+)-ATPase is located at the basolateral membrane complex attached to actin cytoskeleton and is essential for the transtubular vectorial reabsorption of sodium. Ischemia or cellular ATP depletion leads to the rapid redistribution of (Na+-K+)-ATPase into the apical membrane domain of the proximal tubule.30 Therefore, following depletion of energy stores, electrolyte (Na, K) gradients collapse and actin-containing microfilaments become disorganized.2
Decrease in urine volume and in free-water clearance indicated water retention in cirrhotic rats with RF. Pathogenesis of water retention in cirrhosis is complex and probably involves several factors, including increased plasma levels of antidiuretic hormone (ADH), decrease in prostaglandin synthesis and reduced delivery of filtrate to the ascending limb of Henle.31,32 It should be emphasized that increased sodium excretion together with water retention led to dilutional hyponatremia. Studies in humans and experimental animals provided strong evidence that ADH plays a major role in the pathogenesis of water retention in cirrhosis. Longitudinal studies in rats with cirrhosis and ascites have shown a chronological relationship between ADH hypersecretion and impaired water excretion.33,34 In addition, kidneys from cirrhotic rats with ascites show increased gene expression of aqua-porin-2, the ADH-regulated water channel.35 In most patients with ascites free-water clearance is reduced.31 Our data are in accordance with this report. Water retention contributes to dilutional hyponatremia observed in patients with advanced liver disease.36,37
Hypoxic injury to tubular cells represents an early event in acute renal failure. Our data showed that both, renal hemodynamics and proximal tubular function were damaged in DC with RF rats. Persistent renal hypoxia may favor occurrence of tubular damage associated to ATP depletion, as shown in this study. In regard to renal vasoconstriction during RF, sympathetic axis can be stimulated by three different mechanisms: a) activation of pressure receptors in response to hypotension, b) non-volume-dependent hepatic baroreceptors and c) secondary to metabolic changes like catecholamines secretion in response to hypoglycemia. All of these mechanisms might be active in RF.4 Hypoglycemia of the RF rats reported in this study is in accordance with the third mechanism related to strong activation of sympathetic nervous system in patients with RF.38
In summary, CCl4-treatment to cirrhotic rats with RF reduces GFR and filtration fraction and produces proximal tubular damage characterized by increased sodium, glucose and phosphate excretion, decreased renal ATP concentration and reduced free-water clearance. Our study provides a model of liver damage similar to what occurs in decompensated cirrhotic patients and brings the possibility to study the overlapping effects of deleterious agents that might induce renal failure in them.



