




Introduction. The early establishment of an etiology for acute liver failure (ALF) in infants is essential for the start of adequate treatment in the shortest timeframe possible. Aim. To identify markers of inherited metabolic disease on admission in children under two years of age with ALF.
Material and methods. A retrospective review of the medical records of all children (< 2 years old) with ALF admitted to the pediatric hepatology or intensive care units of a tertiary center over a twenty-three year period (January 1989 to December 2011) was done. Patients were divided into two groups: with (group A) or without (group B) a metabolic etiology. Clinical and laboratory parameters on admission were compared.
Results. Twenty-three children met inclusion criteria. Twelve had ALF of metabolic origin (group A). The median age in this group was 2.25 (Q1-Q3: 0.63-4.65) months and in group B 8.0 (Q1-Q3: 1.5-15) months. History of failure to thrive and/or vomiting was more frequent in group A (p = 0.022). Age, gender, encephalopathy and left ventricular hypertrophy were similar in both groups (p = 0.147, p = 1.000, p = 0.637, p = 1.000, respectively). Laboratory tests on admission (plasma lactate, ammonia, cholesterol, phosphate, INR, glucose, bilirubin, ALT, base excess and the presence of reducing substances in urine) showed no statistically significant differences between groups.
Conclusion. This study showed that although infants with inborn errors of metabolism showed a trend towards lower age at presentation, the only marker of inherited metabolic disease found on admission was history of vomiting and/or failure to thrive.
Acute liver failure (ALF) in children is a rare mul-tisystemic disorder with high morbidity and mortality. It is defined as severe impairment of liver function, with or without encephalopathy, in association with hepatocellular necrosis, in a patient with no recognized underlying chronic liver disease.1
In young children, the identification of encephalopathy is not mandatory for the definition of ALF, since its signs are difficult to identify and encephalopathy can be a late event in the course of the disease.2 INR, which expresses prothrombin time, is very helpful in this age group and can be used as an indicator of the severity of liver damage, thus helping decide whether or not to list for transplantation.2
In infants, ALF has several etiologies, the most common of which are liver based inborn errors of metabolism (IEM) and perinatally acquired infections. The former can be responsible for up to 40% of cases.3 Management is directed at treating and monitoring ALF, whilst anticipating its multisystemic complications.4 The diagnosis of the underlying disorder may lead to clinical improvement when appropriate treatment is initiated promptly.5
Galactosemia, tyrosinemia type 1, hereditary fructose intolerance and mitochondrial respiratory chain disorders are the most common IEM associated with ALF.6–8 Patients affected by the first three conditions can improve dramatically with appropriate therapeutic intervention.1 When ALF is severe, liver transplantation is the only life-saving option.2 On the other hand, some IEM are primarily multisystemic disorders and this can be considered a contraindication for liver transplant.4,9
The number of patients presenting with ALF as a first manifestation of metabolic disease has declined in the last few years’ due to earlier diagnosis of liver metabolic diseases. Neonatal screening, implemented in Portugal in 1979 for phenylketonuria, was recently extended, allowing earlier recognition of liver involvement in neonates.
The aim of this study is to identify markers of metabolic disease on admission in children under two years of age with ALF.
Material and MethodsA descriptive retrospective review of the medical records of all children (< 2 years old) with ALF admitted to the pediatric hepatology or intensive care units of a tertiary center over a 23-year period (January 1989 to December 2011) was made.
ALF was defined as coagulopathy (INR > 2) unresponsive to vitamin K, regardless of clinical hepatic encephalopathy or neurological abnormalities. Infants with evidence of chronic liver disease at presentation were excluded, as well as those without a final etiological diagnosis.
The initial baseline investigation in a child with ALF included: full blood count and blood smear, creatinine, urea and electrolytes, ammonia, lactacte, glucose, markers of liver synthesis (prothrombin time, INR, factors V and VII, fibrinogen, albumin, cholesterol), markers of necrosis (AST, ALT, LDH), markers of bile duct injury (ALP, GGT, bilirubin), venous blood pH and gases, base excess and presence of reducing substances in urine, abdominal ultrasound, echocardiogram and electroencephalogram.
A diagnostic algorithm was used to establish the etiology of ALF, including the following exams in children < 2 years of age: routine metabolic investigation (neonatal screening for metabolic disease, plasma and urine amino acid chromatography), serum ferritin levels, transferrin saturation, triglycerides, viral markers (herpes simplex virus, enterovirus, adenovirus, herpes virus type 6, varicella zoster, parvovirus B19, Epstein-Barr virus) and DNA extraction. When family history was present, autoimmune markers (ANA, ASMA, LKM, immunoglobulins, direct antiglobulin test) were also included. According to clinical history and exam results, further tests were requested on a child-to-child basis.
Each patient included in the study group was screened with respect to initial presentation and laboratory results. Several items were analyzed: age, gender, history of vomiting or failure to thrive, encephalopathy and family history of liver disease with onset in childhood. The presence of left ventricular hypertrophy was assessed by echocardiogram. Initial laboratory results were collected: lactate, ammonia, cholesterol, phosphate, INR, glucose, bilirubin, ALT, base excess and presence of reducing substances in urine. Patient’s follow-up was briefly described regarding orthotopic liver transplantation and whether death occurred due to ALF and its complications.
The standard classification of hepatic encephalopathy adapted to infants under 4 years of age was used,10 although the different grades of severity are not discriminated in this study. Since this classification cannot be applied to newborns, neurological distress was defined by irritability, poor sucking, inappropriate agitation or crying or a disturbed sleep-wake cycle in this age group.11
Left ventricular hypertrophy was defined as an increase in left ventricular mass (e.g.: ventricular septum and left ventricular posterior wall thickness, according to the child’s age).
According to the etiology of ALF, two groups were defined: Group A with metabolic disease and Group B with other etiologies for ALF. These two groups were compared regarding initial presentation, laboratory data and imaging results.
Statistical analysis was performed using the Statistical Package for the Social Sciences version 19.0 (SPSS Inc., Chicago, IL). The description of the population was made by calculating measures of central tendency and dispersion for quantitative variables and by determination of absolute and relative frequencies for qualitative variables. The MannWhitney U test was used to compare quantitative variables without normal distribution. The Fisher exact test was used for the association between two qualitative variables, as the lowest expected frequency was below 5. The Kaplan-Meier method was used to estimate in both groups the requirement for liver transplantation and the survival curve. The statistical significance level was set at p < 0.050.
ResultsDuring the 23 years of this study a total of 54 patients were admitted to Hospital Pediátrico de Coimbra with ALF, 28 of whom were < 2 years of age. Out of these, 5 (18%) were excluded from the study, as ALF etiology remains unknown. A total of 23 children met inclusion criteria, of which half (n = 12) were included in group A (with metabolic disease) and 11 in group B (without metabolic disease).
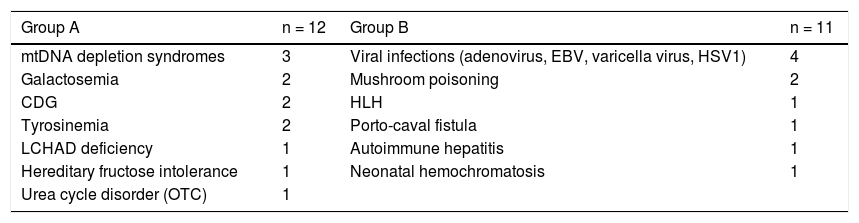
The etiologies of ALF in the study group are shown in table 1. In the inherited metabolic diseases group (A) the most common disorders were mitochondrial DNA depletion syndromes (3 cases, 2 with deoxyguanosine kinase - DGUOK - deficiency), followed by tyrosinemia type 1, galactosemia and congenital disorders of glycosylation type X (2 each). In group B, which includes all other etiologies, viral infection was the most common diagnosis (4 patients), followed by mushroom poisoning and hemophagocytic lymphohistiocytosis (2 each).
Etiology groups.
| Group A | n = 12 | Group B | n = 11 |
|---|---|---|---|
| mtDNA depletion syndromes | 3 | Viral infections (adenovirus, EBV, varicella virus, HSV1) | 4 |
| Galactosemia | 2 | Mushroom poisoning | 2 |
| CDG | 2 | HLH | 1 |
| Tyrosinemia | 2 | Porto-caval fistula | 1 |
| LCHAD deficiency | 1 | Autoimmune hepatitis | 1 |
| Hereditary fructose intolerance | 1 | Neonatal hemochromatosis | 1 |
| Urea cycle disorder (OTC) | 1 |
CDG: congenital disorders of glycosylation. EBV: Epstein-Barr virus. HLH: hemophagocytic lymphohistiocytosis. HSV1: herpes simplex virus 1. LCHAD: long-chain acyl CoA dehydrogenase. mtDNA: mitochondrial DNA. OTC: ornithine transcarbamylase.
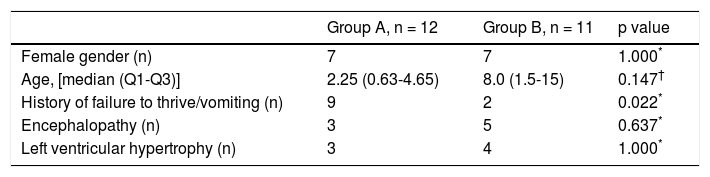
Male/female ratio was similar in both groups (Table 2). Median age of presentation was 2.25 (Q1-Q3: 0.634.65) months in group A, and 8.0 (Q1-Q3: 1.5-15) months in group B, with no statistical significance (p = 0.147) (Table 2). The youngest infant presented at 2 days of life and was diagnosed with neonatal hemochromatosis.
Clinical and diagnostic features.
| Group A, n = 12 | Group B, n = 11 | p value | |
|---|---|---|---|
| Female gender (n) | 7 | 7 | 1.000* |
| Age, [median (Q1-Q3)] | 2.25 (0.63-4.65) | 8.0 (1.5-15) | 0.147† |
| History of failure to thrive/vomiting (n) | 9 | 2 | 0.022* |
| Encephalopathy (n) | 3 | 5 | 0.637* |
| Left ventricular hypertrophy (n) | 3 | 4 | 1.000* |
n: number of patients. Q1: quartile 1. Q3: quartile 3.
Family history was irrelevant in all cases except in one DGUOK patient whose brother had died in another hospital with the diagnosis of neonatal hemochromatosis, later proved to be also a DGUOK deficiency.
Regarding past medical history, 9 patients in group A (82%) and 2 patients in group B (18%) presented with vomiting episodes and/or failure to thrive (p = 0.022). The presence of encephalopathy was also compared (3 in group A and 5 in group B), with no statistical significance (p = 0.637). A similar number of patients presented with left ventricular hypertrophy in both groups (p = 1.00) (Table 2).
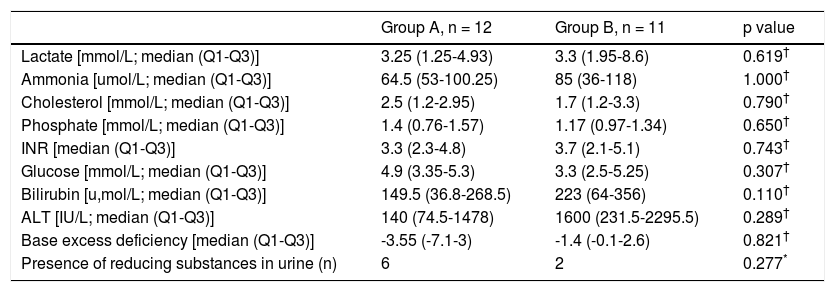
The mean or median levels of each laboratory value analyzed (lactate, ammonia, cholesterol, phosphate, INR, glucose, bilirubin, ALT and base excess) and the number of patients with presence of reducing substances in urine in both groups are shown in table 3. No statistically significant differences were found in any of these parameters.
Laboratory data evaluated on admission.
| Group A, n = 12 | Group B, n = 11 | p value | |
|---|---|---|---|
| Lactate [mmol/L; median (Q1-Q3)] | 3.25 (1.25-4.93) | 3.3 (1.95-8.6) | 0.619† |
| Ammonia [umol/L; median (Q1-Q3)] | 64.5 (53-100.25) | 85 (36-118) | 1.000† |
| Cholesterol [mmol/L; median (Q1-Q3)] | 2.5 (1.2-2.95) | 1.7 (1.2-3.3) | 0.790† |
| Phosphate [mmol/L; median (Q1-Q3)] | 1.4 (0.76-1.57) | 1.17 (0.97-1.34) | 0.650† |
| INR [median (Q1-Q3)] | 3.3 (2.3-4.8) | 3.7 (2.1-5.1) | 0.743† |
| Glucose [mmol/L; median (Q1-Q3)] | 4.9 (3.35-5.3) | 3.3 (2.5-5.25) | 0.307† |
| Bilirubin [u,mol/L; median (Q1-Q3)] | 149.5 (36.8-268.5) | 223 (64-356) | 0.110† |
| ALT [IU/L; median (Q1-Q3)] | 140 (74.5-1478) | 1600 (231.5-2295.5) | 0.289† |
| Base excess deficiency [median (Q1-Q3)] | -3.55 (-7.1-3) | -1.4 (-0.1-2.6) | 0.821† |
| Presence of reducing substances in urine (n) | 6 | 2 | 0.277* |
n: number of patients. Q1: quartile 1. Q3: quartile 3.
In the study group, 4 children, with the following disorders, were submitted to an orthotropic liver transplant: mtDNA depletion syndrome, varicella virus infection, mushroom poisoning and autoimmune hepatitis. Figure 1A shows the proportion of patients without liver transplantation in both groups.

A total of 9 children died due to ALF and its complications, 3 in group A (2 mtDNA depletion syndromes, 1 LCHAD deficiency) and 6 in group B (2 viral infections, 2 hemophagocytic lymphohistiocytosis, 1 porto-caval fistula, 1 autoimmune hepatitis) (p = 0.214). Out of these, 4 had contraindications for liver transplantation. Most children (n = 7) died from multiple organ failure, 1 from severe brain hemorrhage and 1 shortly after liver transplantation (autoimmune hepatitis). Figure 1B shows the survival curve in both groups.
DiscussionIn the present study the etiology for ALF was determined in 23 of 28 patients under 2 years of age, with only 18% remaining without a diagnosis. Another study, which reviewed patients with ALF younger than 1 year of age, had similar results, with 16% of cases with unknown diagnosis.11 This is in contrast with two series of ALF, one in infants aged ≤ 90 days12 and another in children from birth to 18 years of age,13 that presented respectively with 38 and 49% of cases with indeterminate diagnosis.
Liver based IEM present frequently in the neonatal period with jaundice, severe hypoglycemia and sometimes with ALF.6 They represent an important cause of ALF in < 2 year olds. Therefore, IEM must always be considered in an infant presenting with ALF, as an appropriate diet or specific treatment might be lifesaving.2 In several pediatric case series of ALF the percentage of IEM as an etiology varied between 10% and 42.5%.11–13 This wide spectrum may be explained not only by the different investigation protocols, but also by the diverse age groups included in each study.
In the present study 43% of the 28 patients < 2 years of age were diagnosed with an inherited metabolic etiology for ALF. A similar percentage (42.5%) was found in a group of ALF patients under one year of age.11 In older children, IEM present more rarely and secondary causes of ALF, namely viral and toxic, became more important.14
The only exception might be neonatal hemochromatosis, an alloimune disease and the most frequently recognized cause of neonatal ALF, which presents typically very early in life.15,16 In this series, which included one patient with neonatal hemochromatosis, children with IEM showed a trend towards lower age on admission [group A 2.25 (Q1-Q3: 0.63-4.65) vs. group B 8.0 (Q1-Q3: 1.5-15) months], although the age difference was not statistically significant.
A specific treatment is available in some IEM with significant liver involvement. Tyrosinemia type 1, galactosemia and fructosemia, three of the most commonly described etiologies, are IEM due to intoxication, amenable to effective dietary/ pharmacological treatment.6,7 In recent years, mitochondrial respiratory chain defects, which are a broad group of diseases, frequently multisystemic, with no specific treatment, have been implicated as an etiological factor for ALF in infants.17
The etiology of group A of the series in discussion was varied, with mitochondrial disorders, tyro-sinemia type 1, galactosemia and congenital disorders of glycosylation representing the most frequent diagnosis. The two cases with congenital disorders of glycosylation have been ascribed an X type, since the diagnosis was based on transferrin isoelectric focusing pattern abnormalities, not explained by liver failure, with no enzyme or genetic diagnosis yet.
In group B, the most frequent diagnosis was a viral infection, as expected.11–13 However, even if an infectious etiology is found, an IEM should be considered in this age group.18
Failure to thrive and/or vomiting have been described as striking features in the clinical presentation of IEM.19 In this study, only this presenting feature proved to be statistically significant between both groups, with a larger number of cases in group A. Thus, its presence should point in the direction of an inherited metabolic disorder, particularly in infants.
Many IEM produce dysfunction in other organs such as the central nervous system and heart, which can be very helpful indicators in their differential diagnosis.20 However, ALF is an important cause of encephalopathy. In the clinical setting, the distinction between the role of liver failure or that of a probable underlying central nervous system disease (e.g.: viral infection, multisystemic IEM) in a child with encephalopathy is very hard. As would be expected in this age group, in this study only a small number of children were described as having encephalopathy. The slightly larger number of patients with encephalopathy in group B (Table 2) might be explained by the older age of children in this group and, therefore, with easier to diagnose mental status changes. Although without statistical significance, this may be a possible study bias.
Heart involvement, namely hypertrophic cardiomyopathy, is a rather common feature in IEM.21 In this study, the number of patients with left ventricular hypertrophy was similar in both groups, contrarily to what was anticipated.
Very little data was found regarding biological markers to aid in differentiating metabolic from non-metabolic disease in children presenting with ALF. A recent study has showed that in a population of 148 infants ≤ 3months of age with ALF there was a moderate association between the cumulative biochemical profile and the presence of ALF in infants with IEM, when compared with neonatal hemochromatosis: patients with IEM had moderately elevated aminotransferase levels, moderate cholestasis, and minimal coagulopathy.12
One of the routine tests for diagnosis of liver IEM, namely galactosemia and fructosemia, is to ascertain the presence of reducing substances in urine,22 and this test is frequently used to guide in identifying the etiology of ALF. In this series, the number of infants with reducing substances in urine was greater in group A, although without statistical significance. Children with hepatic failure, regardless of their etiology, may have reducing substances in urine as liver metabolic functions are secondarily impaired. Therefore, it is important not to allow this test to supersede the search for other possible etiologies for ALF other than IEM.
In this study, bilirubin and ALT showed a trend towards higher levels in group B. Unexpectedly, plasma glucose levels were lower in group B than in group A. None of these differences or those of the other markers also mentioned in table 3 were statistically significant, thus not permitting a more direct approach in diagnosing the etiology of ALF. As a result, specific laboratory markers for each disease are still the mainstay for diagnosis.
In conclusion, ALF is an important cause of morbidity and mortality particularly in under two year-old children, and early establishment of an etiology is warranted. In this series, history of vomiting and/ or failure to thrive was shown to be a possible marker of IEM on admission in this age group.
A possible bias of this study is the small number of cases presented, which does not allow to make definitive conclusions. Hence, more studies on this issue should be undertaken, with larger cohorts of patients, to understand if the infants’ clinical and laboratory profile may help to distinguish between a probable IEM and a non-metabolic cause for ALF. This would be helpful in the management of these patients, namely in the decision making of listing for liver transplantation.
Abbreviations- •
ALF: acute liver failure.
- •
ALP: alkaline phosphatase.
- •
ALT: alanine aminotransferase.
- •
ANA: antinuclear antibodies.
- •
ASMA: anti-smooth muscle antibodies.
- •
AST: aspartate aminotransferase.
- •
DGUOK: deoxyguanosine kinase.
- •
GGT: gamma-glutamyl transpeptidase.
- •
INR: international Normalized Ratio.
- •
IEM: inborn errors of metabolism.
- •
LDH: lactate dehydrogenase.
- •
LKM: liver/kidney microsomal antibody.
None.



