




Over the past three decades, there has been a remarkable improvement in the outcome of children diagnosed with systemic lupus erythematosus (SLE). In general, paediatric-onset SLE has been associated with higher mortality rates and more disease damage than adults with SLE. The objective was to determinate the impact of clinical, laboratory, and electroencephalographic findings on survival amongst patients with paediatric-onset SLE.
MethodsCharts of Mexican patients with paediatric-onset SLE diagnosed between 1970 and 2001 were analysed retrospectively; univariate and multivariate analyses were used for analysing associations between clinical and laboratory features and death; Kaplan–Meier tests were used to estimate survival curves.
Results159 patients were included, 105 were female, with a median age of 12.7 years at diagnosis and a median duration of symptoms prior to diagnosis of 8.4 months. Univariate analysis showed that haematuria, leukocyturia, proteinuria, presence of urine cast, <60% glomerular filtration rate, haemolytic anaemia, and abnormal electroencephalogram, were all poor prognostic factors (p<0.05). Multivariate analysis showed that the presence of proteinuria and abnormal electroencephalograms (p<0.05) were independent factors associated with death. The overall survival rate was 82.9% at five years and 77.4% at ten years upon follow-up. Infection and high disease activity were the most common causes of death.
ConclusionsSurvival of paediatric-onset SLE patients was lower compared to that reported for patients in wealthier countries. Amongst the patients who died, the presence of proteinuria and abnormal electroencephalograms were found to be determinant for survival. Infection and activity were the most common causes of death.
Systemic lupus erythematosus (SLE) is an autoimmune disease that predominantly affects females,1 and 15–20% of all SLE patients are diagnosed before the age of 16. Over the past three decades, there has been a remarkable improvement in the outcome of children diagnosed with SLE.2–5 In general, paediatric-onset SLE has been associated with higher mortality rates and more disease damage than adults with SLE.6,7 The presence of lupus nephritis in children is associated with a higher mortality risk.8,9
The survival rate of paediatric-onset SLE patients in Mexico is currently unknown. The objectives of this retrospective study were to evaluate the prognostic value of clinical, laboratory, and electroencephalographic findings as well as to examine long-term survival in Mexican patients with paediatric-onset SLE.
Patients and methodsMedical records of Mexican patients diagnosed between 1970 and 2001 who fulfilled the revised criteria from the American College of Rheumatology (ACR, 1982) for diagnosis of SLE before the age of 18 at the Immunology Department of the “Instituto Nacional de Pediatría” (National Institute of Paediatrics) were retrospectively reviewed.10 The centre is a highly specialised paediatric hospital for children located in Mexico City and receives patients from all over the country, particularly with low socioeconomic status and lack of medical insurance. Most patients received their last follow-up appointment at the age of 18, although some received follow-ups until 26 years of age due to hospital policy. All patients admitted to the immunology department were systematically evaluated through clinical, laboratory and ancillary evaluations to rule out organ failure even though there was no evidence of clinical affection; assessment on neuropsychiatric, haematologic, ocular, renal, cardiovascular, liver and pulmonary systems were carrying out.
A data collection form was used to concentrate information about demographic data, clinical manifestations, laboratory findings, and electroencephalographic reports at disease onset.
Demographic data included gender, age at onset of disease (defined as the age at which the first symptoms clearly attributable to SLE occurred), age at diagnosis (defined as the age when patients fulfilled diagnosis criteria for SLE), follow-up time (defined as the time from disease onset until the last visit, or death), and diagnostic delay (defined as time between onset of disease and diagnosis). Clinical and laboratory data were defined by the following criteria: proteinuria (≥0.5g/day), leukocyturia in the absence of infection (≥5 leukocytes per high-powered field), haematuria (≥5 red blood cells per high-powered field), the presence of cellular casts of any type, <60% glomerular filtration rate, haemolytic anaemia (anaemia and positive Coombs’ test), and thrombocytopenia (platelets <100,000/mm3); the neuropsychiatric lupus syndromes were defined using the 1999 ACR nomenclature, including the case definition for the 19 different kinds.11
Patients who died were recorded; information on the causes of patient death was obtained from the clinical files and death certificates. Death was considered to be due to active SLE if related to uncontrollable and progressing SLE. Infections were diagnosed on clinical grounds and confirmed by other available techniques as cultures.
Patients were excluded from the study if they had drug-induced SLE, isolated discoid lupus, mixed connective tissue disease, had received prior SLE diagnosis and/or treatment in another medical centre, or missed follow-up appointments during the first year of disease. The study protocol was approved by the Institutional Review Board of the National Institute of Pediatrics.
Statistical analysisStatistical Package for Social Science (SPSS) version 12.0 was used for data analysis. Results are presented as mean±standard deviation if they have a normal distribution and median (interquartile range) for continuous variables and as percentages for nominal variables if they have not a normal distribution. Univariate analysis was performed by an independent sample t-test to compare the means, and differences were assessed by Pearson's chi-square test. The probability curves of survival were calculated by the Kaplan–Meier method and compared by the log-rank test. Multivariate analysis was performed using the Cox proportional hazard model to identify an independent risk factor for poor survival. All two-sided P-values<0.05 were considered to be statistically significant.
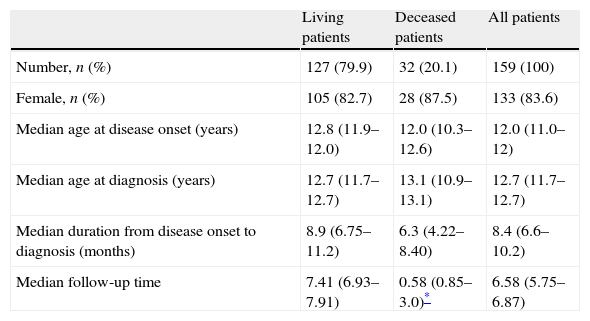
ResultsDemographic dataA total of 169 patients fulfilled the ACR classification criteria to be included in our study. Ten patients (5.2%) were lost to follow-up during the first year and were therefore excluded from our analysis. Demographic data are summarised in Table 1. Mean age at onset of disease in males (11.5±.2 years) was almost identical to that observed in females (11.5±2.9 years). Female to male ratio was 5.1:1. Fifty-four patients (34%) started with signs and symptoms of SLE at the age of 10 years or younger, and 105 (66.0%) were 11 years or older (Table 1).
Demographic information of the 159 SLE patients in our study.
| Living patients | Deceased patients | All patients | |
| Number, n (%) | 127 (79.9) | 32 (20.1) | 159 (100) |
| Female, n (%) | 105 (82.7) | 28 (87.5) | 133 (83.6) |
| Median age at disease onset (years) | 12.8 (11.9–12.0) | 12.0 (10.3–12.6) | 12.0 (11.0–12) |
| Median age at diagnosis (years) | 12.7 (11.7–12.7) | 13.1 (10.9–13.1) | 12.7 (11.7–12.7) |
| Median duration from disease onset to diagnosis (months) | 8.9 (6.75–11.2) | 6.3 (4.22–8.40) | 8.4 (6.6–10.2) |
| Median follow-up time | 7.41 (6.93–7.91) | 0.58 (0.85–3.0)* | 6.58 (5.75–6.87) |
a Median (interquartile range).
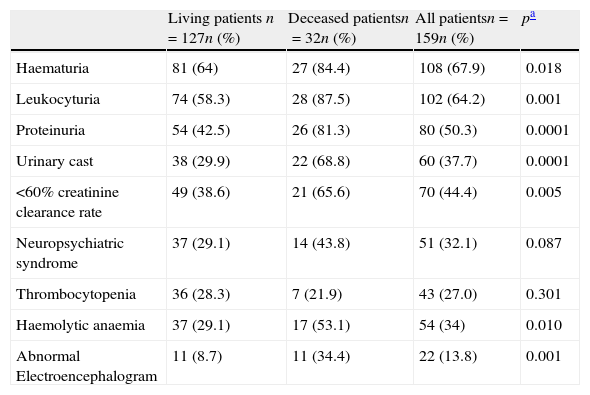
Prevalence of clinical, laboratory and EEG abnormalities at disease onset are summarised in Table 2. Laboratory abnormalities reported at diagnosis were mainly related to renal disease. Neuropsychiatric syndromes related to SLE were most frequently diagnosed in our group of patients. Amongst the case definition for the 19 different kinds of neuropsychiatric lupus syndromes the most prevalent were cognitive dysfunction (n=4), seizures (n=19), and cerebrovascular disease (n=6). All patients had at least one electroencephalogram at diagnosis. Only eight of 22 patients (36%) with an abnormal EEG had clinically evident seizures.
Clinical, laboratory and EEG features at disease diagnosis in 159 SLE patients.
| Living patients n=127n (%) | Deceased patientsn=32n (%) | All patientsn=159n (%) | pa | |
| Haematuria | 81 (64) | 27 (84.4) | 108 (67.9) | 0.018 |
| Leukocyturia | 74 (58.3) | 28 (87.5) | 102 (64.2) | 0.001 |
| Proteinuria | 54 (42.5) | 26 (81.3) | 80 (50.3) | 0.0001 |
| Urinary cast | 38 (29.9) | 22 (68.8) | 60 (37.7) | 0.0001 |
| <60% creatinine clearance rate | 49 (38.6) | 21 (65.6) | 70 (44.4) | 0.005 |
| Neuropsychiatric syndrome | 37 (29.1) | 14 (43.8) | 51 (32.1) | 0.087 |
| Thrombocytopenia | 36 (28.3) | 7 (21.9) | 43 (27.0) | 0.301 |
| Haemolytic anaemia | 37 (29.1) | 17 (53.1) | 54 (34) | 0.010 |
| Abnormal Electroencephalogram | 11 (8.7) | 11 (34.4) | 22 (13.8) | 0.001 |
The overall survival rate was 82.9% at five years and 77.4% after a 10-year follow-up (Fig. 1). There were a total of 32 deaths (20.1%) during the follow-up period with a mean age at the time of death of 13.9±2.28 years; the median time of death after diagnosis was seven months (range: 0–148).
. The vertical ticks represent censored patients.")
The causes of death were classified as related to infection associated with activity in 15 patients (46.9%); disease activity in nine (28.1%); infection in seven (21.9%); and M4 acute myelomonocytic leukaemia associated with infection in one patient (3.1%). Twenty-one patients (57.1%) died during the first year of follow-up treatment primarily due to infection associated with an active disease (n=12).
The reported causes of death amongst patients with high disease activity were acute myocardial infarction (n=2); pulmonary haemorrhage (n=1); stroke (n=2); cerebral vasculitis (n=1); severe hypertension (n=1); status epilepticus (n=1); and acute renal failure (n=1).
Amongst patients who died because of infection, sepsis with primary pulmonary focus was the main cause of death (n=22). Microorganisms were isolated in very few cases and were (one for each agent): beta-haemolytic streptococci, Burkholdelia cepacia, Enterobacter aerogenes, Nocardia sp., Aspergillus sp., and Histoplasma sp.
An autopsy examination was performed in only one patient and revealed bacterial endocarditis and anatomical findings related to septic shock.
The ten-year survival rate was significantly affected by the presence at disease onset of haematuria, leukocyturia, proteinuria, <60% glomerularfiltration rate, urine casts, an abnormal EEG, or haemolytic anaemia at diagnosis in the univariate analysis (Fig. 2).
 Probability of survival at ten years in SLE patients at disease onset with and without leukocyturia; (b) with and without proteinuria; (c) with and without urine cast; (d) with and without EEG.")
Using a multivariate analysis, the presence of an abnormal EEG (p=0.0001; OR=4.6; 95% CI=2.2–9.7) and proteinuria (p=0.0001; OR=5.0; 95% CI=2.0–12.7) were independent factors associated with the risk of death.
DiscussionThere are several publications regarding SLE survival from all over the world that report different survival and mortality rates which are influenced by ethnic, socioeconomic, educational and demographic factors.12,13 Our results showed that in the majority of the patients, SLE was diagnosed after the age of 10 years, in accordance with various series of children and adolescents with SLE.12
The patients in our study had a longer delay in diagnosis compared to those from wealthier countries.13 Swaak et al. previously described that in 110 patients, the time between disease onset and diagnosis was longer amongst patients who died than those who survived.14
A marked improvement in survival rates around the world, over time amongst patients with SLE has been documented and it is attributed to an earlier recognition of the disease and better therapeutic approaches.4 Several studies reported five- and ten-year survival rates ranging from 61% to 100% and from 28% to 90% respectively.5,13 The survival rate amongst Mexican patients with paediatric-onset SLE was lower in comparison to that reported for patients from wealthier countries.13,15 Nonetheless, survival rates were similar or greater in comparison to developing countries.16 Economic condition and ethnic factors could explain the differences. Low income and poor education are often associated with greater difficulties in accessing to health care, inability to understand and recognise SLE symptoms, inability to carry out medical recommendations, and inadequate social support, nutrition, and employment opportunities.17 The roll of ethnic factors in SLE prognosis is controversial.19 Pons-Estel et al. showed that patients of Latin American origin had poor prognosis.18
Amongst the diverse SLE manifestations at disease onset we decided to analyse those previously associated with poor prognosis as renal and neuropsychiatric affection, haemolytic anaemia and thrombocytopenia. As described previously, renal disease is more common in paediatric-onset SLE.19 In our patients, renal damage was the most commonly affected organ.2,22,23 The most frequent neuropsychiatric lupus syndromes (defined using the ACR nomenclature) in our group of patients included cognitive dysfunction, seizures and cerebrovascular disease as reported previously by Yu et al. in 2006.20We confirmed, as other reports that haemolytic anaemia, renal or abnormal EEG at onset of disease were also associated to a poor prognosis.1,6,9,19–25
In our hospital, since 1970 regardless of the presence of seizures at onset of disease, all patients suspected of having SLE initially had an electroencephalogram as a part of screening of subtle neurological damage.
Abnormal EEG at onset disease in patients was associated to a poor prognosis. EEGs are central to the diagnosis and management of epilepsy but can also help in the diagnosis and prognosis of diffuse encephalopathies, organic brain syndromes, and dementias,26 which have been associated with underlying vascular changes in the brain of SLE patients secondary to antiphospholipid syndrome and antiphospholipid antibodies.20,26 EEG results in our patients are interesting since this is not something that many centres do routinely. We suggest that EEG should always be performed even without obvious neurological symptoms because many abnormalities could be found even in asymptomatic patients. However, it is an area to be explored further in the future.
Infection and disease activity were the primary causes of death in most patients. In patients with infection, sepsis associated to pneumonia was the principal cause of death. Infection continues to be a leading cause of morbidity and mortality amongst SLE patients.27 As infection (sometime undiagnosed) may trigger disease activity and vice versa, it is frequent to have difficulties to differentiate between both problems.
In paediatric-onset SLE the patient must be closely monitored and aggressively treated to improve survival rates, in particular patients with a disease onset associated to renal damage and an abnormal EEG.
ConclusionsWe summarise the clinical features of 159 paediatric patients with SLE evaluated at the National Institute of Paediatrics between 1970 and 2001. We conclude that clinical findings frequently reported at diagnosis were more often related to renal involvement. In fact, renal disease, haemolytic anaemia, and having an abnormal electroencephalogram at the onset of disease were all associated with a poor prognosis. The overall survival rate of patients was 82.9% at five years and 77.4% at ten years upon follow-up treatment. The cause of death in most of the patients was high disease activity or infection during the first few months after diagnosis.
Conflict of interestThe authors declare that they have no conflicts of interest.
We would like to thank Chiharu Murata, M.Sc., for help with statistical analysis.
