



El síndrome de Sjögren primario (SSP) es una enfermedad autoinmune sistémica que se caracteriza por una infiltración linfocitaria en las glándulas exocrinas, principalmente las glándulas salivales y lagrimales, que produce xeroftalmia y xerostomía. En ocasiones cursa con manifestaciones extraglandulares, incluyendo el sistema nervioso central (SNC) y el periférico (SNP). La prevalencia de estas entidades es variable en la literatura, debido principalmente a la heterogeneidad de criterios diagnósticos y clasificatorios. La afectación del SNP es más frecuente, y puede presentarse como neuropatías axonales, sensitivas, autonómicas, de fibra pequeña y mononeuritis múltiple. Las manifestaciones del SNC pueden ser focales o difusas, y se asocian con una mayor afectación extraglandular y autoanticuerpos. El tratamiento de estas entidades se basa en el uso de glucocorticoides. En casos graves refractarios o cortico-dependientes pueden utilizarse inmunosupresores como la azatioprina o la ciclofosfamida. Algunos estudios observacionales sugieren que rituximab es eficaz en ciertas manifestaciones extraglandulares del SSP. Actualmente se están llevando a cabo ensayos clínicos con nuevos agentes biológicos, como belimumab y epratuzumab.
Primary Sjögren syndrome (PSS) is an autoimmune disease characterized by lymphocytic infiltration in exocrine glands, mainly the salivary and lachrymal glands, resulting in xerophthalmia and xerostomia. Occasionally, PSS has extra-glandular manifestations, including central (CNS) and peripheral nervous system (PNS) involvement. The reported prevalence varies in the literature, mainly because of heterogeneity in the diagnostic and classification criteria. Involvement of the PNS is more frequent than that of the CNS, presenting as axonal, sensory, autonomic or small fiber neuropathies and mononeuritis multiplex. CNS manifestations may be focal or diffuse and are associated with increased extra-glandular involvement and the presence of autoantibodies. Treatment is based on corticosteroids. In severe, refractory- or corticosteroid-dependant cases, immunosuppressant agents such as azathioprine or cyclophosphamide may be useful. Observational studies have suggested that rituximab is useful in some PSS extra-glandular manifestations. Currently, clinical trials with new biological agents such as belimumab and epratuzumab are being conducted in PSS.
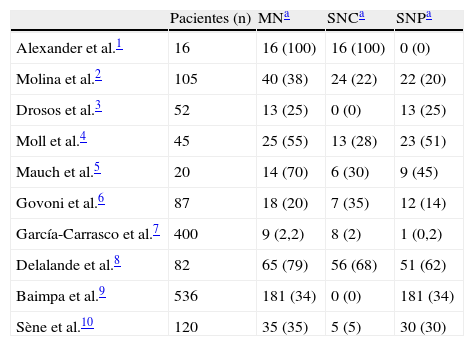
El síndrome de Sjögren primario (SSP) es una enfermedad autoinmune sistémica caracterizada por una infiltración linfocitaria de las glándulas exocrinas, principalmente salivales y lagrimales. Como consecuencia se produce una destrucción glandular progresiva y una disminución de la función secretora que conduce a la expresión clínica característica de xeroftalmia y xerostomía. Su prevalencia oscila entre el 0,2 y el 3% y afecta predominantemente a mujeres, con una media de edad entre 40 y 50años. Desde la descripción inicial por Sjögren en 1935 se han definido ampliamente las manifestaciones neurológicas asociadas, incluyendo el sistema nervioso central (SNC) y el periférico (SNP). Como otras manifestaciones extraglandulares de la enfermedad, la prevalencia de alteraciones neurológicas resulta controvertida, oscilando entre el 8,5 y el 70%1-12 (tabla 1 y fig. 1). Se estima que hasta en el 30 al 50% de los casos coexisten las manifestaciones centrales y periféricas1,8. Esta discrepancia en cuanto a prevalencia se encuentra parcialmente explicada por la variabilidad de criterios diagnósticos, por la subespecialidad que atiende al paciente y por la realización sistemática de pruebas complementarias a pacientes asintomáticos.
Prevalencia de manifestaciones neurológicas en el síndrome de Sjögren primario
| Pacientes (n) | MNa | SNCa | SNPa | |
| Alexander et al.1 | 16 | 16 (100) | 16 (100) | 0 (0) |
| Molina et al.2 | 105 | 40 (38) | 24 (22) | 22 (20) |
| Drosos et al.3 | 52 | 13 (25) | 0 (0) | 13 (25) |
| Moll et al.4 | 45 | 25 (55) | 13 (28) | 23 (51) |
| Mauch et al.5 | 20 | 14 (70) | 6 (30) | 9 (45) |
| Govoni et al.6 | 87 | 18 (20) | 7 (35) | 12 (14) |
| García-Carrasco et al.7 | 400 | 9 (2,2) | 8 (2) | 1 (0,2) |
| Delalande et al.8 | 82 | 65 (79) | 56 (68) | 51 (62) |
| Baimpa et al.9 | 536 | 181 (34) | 0 (0) | 181 (34) |
| Sène et al.10 | 120 | 35 (35) | 5 (5) | 30 (30) |
MN: manifestaciones neurológicas; SNC: sistema nervioso central; SNP: sistema nervioso periférico.

Los datos publicados sobre la afectación del SNP parecen ser más uniformes. No obstante, ante la falta de criterios unificados en cuanto al diagnóstico y a la nomenclatura de las neuropatías periféricas, resulta difícil establecer la prevalencia de cada una de las entidades. El diagnóstico de estas manifestaciones es a menudo difícil, con un retraso diagnóstico prolongado13. De hecho, las manifestaciones neurológicas pueden ser la primera manifestación de un SSP e incluso preceder al síndrome seco. La afectación del SNP se asocia más a menudo a xeroftalmia y xerostomía, así como a otras manifestaciones sistémicas, mientras que la vasculitis cutánea parece ser particularmente frecuente en pacientes con afectación del SNC1,2. En la serie de Delalande et al.8 hasta el 47% de los pacientes con manifestaciones neurológicas del SSP no presentaban al inicio de la enfermedad un síndrome seco, especialmente en aquellos con afectación del SNC. Este hecho enfatiza la dificultad en el diagnóstico y en la necesidad de un cribado para SSP en pacientes mayores de 50años con un comienzo de manifestaciones neurológicas, como una polineuropatía sensorial, una mielopatía crónica o una afectación multifocal del SNC.
EtiopatogeniaSe trata de una entidad cuyos mecanismos patogénicos se encuentran mal definidos. En las descripciones histopatológicas procedentes de necropsias y biopsia de nervio periférico14 se encuentra un infiltrado linfocítico en la pared de los vasos de pequeño calibre, constituyendo una vasculitis franca. Este hecho pone de manifiesto el rol de la inmunidad celular por toxicidad directa de los linfocitos T. Asimismo se ha observado una disminución tanto del número total de neuronas de los ganglios sensitivos y simpáticos como de la densidad de fibras mielinizadas en la médula espinal. En las biopsias de nervio periférico se observa una desmielinización parcheada multifocal que a su vez mantiene el diámetro de las fibras, revelando por tanto una degeneración axonal.
Los autoanticuerpos15-17 contribuyen potencialmente a través de diversos mecanismos, incluyendo la vasculitis mediada por inmunocomplejos y la afectación directa sobre la mielina. Las proteínas Ro y La se encuentran presentes en las neuronas, por lo que la presencia de anti-Ro/La en pacientes con afectación neurológica constituye un argumento a favor de su papel en la patogenia. Asimismo, en varios estudios8,13 se ha asociado la presencia de anticuerpos anti-SSA con las manifestaciones neurológicas más graves. De la misma forma, las similitudes entre la neuropatía sensitiva y la paraneoplásica postulan un origen fisiopatológico común, con la producción de anticuerpos antineuronales que simulan anti-Hu, ya que el antígeno Ro presenta epítopos comunes con el antígeno Hu neuronal. Por otro lado, en pacientes en los que predomina la neuropatía autonómica y el síndrome seco se ha evidenciado un aumento de los niveles de anticuerpos anti- receptor de acetilcolina.
Afectación del sistema nervioso periféricoLas neuropatías sensitivomotoras representan las neuropatías periféricas más frecuentes, con una prevalencia del 20 al 30%8 en los pacientes con SSP y manifestaciones neurológicas. La evolución generalmente es lenta e insidiosa. En la mayoría de los casos el inicio se caracteriza por una afectación sensitiva en los miembros inferiores. Es frecuente que la afectación sea distal y simétrica. Los signos motores son, en general, discretos. Hasta en el 15% de los casos puede ser asintomática y ser diagnosticada por un electromiograma (EMG) realizado de forma sistemática. Las alteraciones electrofisiológicas se caracterizan por un patrón axonal que en ocasiones se asocia a denervación. La biopsia de nervio periférico revela adelgazamiento de las fibras mielinizadas y degeneración axonal sin vasculitis asociada.
Entre las neuropatías sensitivas, las ganglionopatías o neuronopatías son las que más frecuentemente se asocian al SSP. Generalmente preceden el diagnóstico de SSP y se asocian en menor medida a otras manifestaciones sistémicas respecto al resto de neuropatías18. Presentan similitudes clínicas y electrofisiológicas con la neuropatía paraneoplásica. Se trata de una entidad que se caracteriza desde el punto de vista anatomopatológico por un infiltrado linfocítico a nivel de los ganglios raquídeos posteriores. Clínicamente los pacientes presentan una ataxia sensitiva19 y parestesias que afectan a todas las extremidades, aunque con predominio en los miembros superiores, y no asocia clínica motora. Los síntomas iniciales suelen ser parestesias en dedos de manos y pies, que frecuentemente son asimétricas y evolucionan hacia una afectación proximal y simétrica. El periodo de instauración suele ser variable, generalmente de meses a años. Algunos pacientes asocian también síntomas autonómicos y disestesias. En la exploración física es característica la marcha atáxica, asociada a una arreflexia generalizada. Puede apreciarse atrofia muscular, así como una maniobra de Romberg positiva. En pacientes con avanzado estado de la enfermedad puede existir incapacidad para la marcha y pueden precisar silla de ruedas. En los estudios neurofisiológicos es característico un patrón axonal asimétrico. Entre los hallazgos en la resonancia magnética (RM) destaca la presencia de un aumento de intensidad de la señal en los cordones posteriores en las secuencias potenciadas en T2. Esta manifestación tiene una buena correlación con la ataxia sensitiva y con la distribución y la intensidad de la afectación sensorial.
La neuropatía de fibra pequeña es una neuropatía prominentemente dolorosa, asociada a reflejos osteotendinosos, sensibilidad vibratoria y estudios de conducción nerviosa normales. Los pacientes realizan una descripción abigarrada de disestesias sin una distribución anatómica correspondiente a un nervio periférico. Presentan zonas parcheadas de hipoestesia nociceptiva y térmica. La mayoría de los casos son indolentes durante meses a años y raramente tienen una presentación aguda. Puede ser una manifestación inicial de SSP y progresar a una alteración profunda en la sensibilidad. La afectación de fibras pequeñas autonómicas es rara. Desde el punto de vista serológico se asocian con mayor frecuencia a anticuerpos antinucleares20. El estudio electromiográfico es normal y los potenciales evocados pueden ser patológicos. El diagnóstico de confirmación se realiza mediante el análisis de fibras nerviosas en una biopsia cutánea, evidenciándose una disminución llamativa de las fibras nerviosas intraepidérmicas. Tanto la prevalencia como la fisiopatología y la evolución clínica son desconocidas. No obstante, algunos autores21 han encontrado una alta prevalencia en cohortes de pacientes con SSP, siendo subclínica en la mayoría de los casos.
Los casos de mononeuritis múltiple asociados al SSP han sido descritos ampliamente en la literatura, aunque se trata de una complicación relativamente poco frecuente. En la serie de Mori et al.14, 11 de 92pacientes (12%) fueron clasificados como mononeuritis múltiple. El síntoma inicial suele ser la aparición brusca de parestesias y disestesias en las porciones distales de los miembros inferiores. Posteriormente los síntomas motores y sensitivos ocurren episódicamente y presentan una distribución restringida a los miembros inferiores. En ocasiones pueden verse afectados el nervio trigémino y los nervios intercostales. Generalmente se asocian afectaciones sistémicas graves, como vasculitis cutánea8 y crioglobulinemia. En el estudio neurofisiológico los potenciales de acción sensitivo y compuesto están marcadamente reducidos. La alteración histológica característica es una vasculitis asociada a necrosis fibrinoide. Tanto la fibra fina como la gruesa pueden encontrarse destacadamente deplecionadas, junto con una degeneración axonal activa.
Son limitados los casos publicados de polirradiculoneuropatía asociados al SSP8,14,22. Clínicamente se define por una afectación sensitivomotora crónica progresiva. Desde el punto de vista sensitivo se caracteriza por parestesias en «guante y calcetín», asociadas o no a ataxia sensitiva. También puede asociarse debilidad muscular. Los síntomas autonómicos generalmente están ausentes, excepto casos aislados de diarrea, hipohidrosis y alteraciones urinarias. El estudio electromiográfico puede revelar conducciones motoras y sensitivas normales junto con anormalidades de la onda F y latencias prolongadas, hallazgos que generalmente son poco frecuentes en una polirradiculoneuropatía desmielinizante crónica. La biopsia de nervio periférico muestra grados variables de pérdida de fibras mielinizadas con cambios leves a moderados en las fibras desmielinizadas.
Si bien hay excepciones, la mayoría de estudios han mostrado que una proporción significativa de pacientes con SSP presenta síntomas y signos de neuropatía autonómica. Estos síntomas pueden observarse asociados a prácticamente todas las formas de neuropatía periférica en el SSP. Respuestas pupilares anormales (pupila de Addie) e hipotensión ortostática son frecuentes en las ganglionopatías pero también pueden presentarse en la neuropatía del nervio trigémino y en la neuropatía de fibra pequeña. En pacientes con hipotensión ortostática grave se ha observado una disminución de la captación en los estudios gammagráficos con I123-metaiodobenzilguanidina14. La base patogénica no está clara: se ha asociado a una ganglionopatía y lesiones vasculíticas en nervios periféricos que transportan fibras autonómicas. La existencia de una pupila de Addie se asocia a una posible neuronitis del ganglio ciliar. Asimismo se han descrito casos de neuropatías autoinmunes autonómicas asociadas a síndrome seco y elevado título de anticuerpos anti-receptor de acetilcolina23.
La afectación de pares craneales más frecuente en el SSP es la del trigémino. En la serie de Mori et al.14, 15 de 92pacientes (17%) presentaban una neuropatía trigeminal sensitiva, 9de ellos unilateral y 6 de forma bilateral. No existen casos descritos de neuropatía motora asociada al nervio trigémino en el SSP. Son características las parestesias correspondientes a las ramas inferiores. Los pacientes también pueden asociar hipoestesias faciales y disestesias en la lengua. Asimismo los pacientes pueden estar asintomáticos, evidenciándose la neuropatía a través de un estudio neurofisiológico sistemático. Para ciertos autores esta entidad puede deberse a una infiltración del ganglio de Gasser19. Pueden afectarse infrecuentemente otros pares craneales, en forma de mononeuropatía craneal múltiple. Es característica la presencia de una paresia facial por afectación del par craneal correspondiente, así como la sordera súbita por alteración del nervio cocleovestibular8. En el caso de una neuropatía de los nervios oculomotores, los pacientes presentan diplopía y estrabismo.
Afectación del sistema nervioso centralLa afectación del SNC en el curso del SSP aparece con menos frecuencia que la del SNP. No obstante, su prevalencia es controvertida, del 1 al 68%. Esta variabilidad se debe en parte a su difícil diagnóstico así como a la inclusión de las manifestaciones psiquiátricas o de las cefaleas en ciertos estudios. Hasta en el 30-50% de los casos coexisten las manifestaciones centrales y periféricas1,8.
Las manifestaciones focales encefálicas son las más frecuentemente observadas. Estas entidades suelen asociarse a fenómenos de vasculitis. Además, el SSP actúa como un factor de riesgo cardiovascular independiente. La inflamación crónica y las características inmunológicas asociadas desempeñan un rol importante en la aterosclerosis acelerada. En un estudio reciente24 en pacientes con SSP se ha asociado un incremento en la aterosclerosis carotídea y femoral con una disregulación en el perfil lipídico, elevados niveles de proteína C reactiva (PCR) y anticuerpos anti-SSA. El inicio de las manifestaciones focales encefálicas suele presentarse de forma aguda como un ictus o un accidente isquémico transitorio. En casos más graves puede existir una hemorragia subaracnoidea. Existe un amplio espectro de manifestaciones clínicas. Entre las más frecuentes8 figuran la hemiparesia, seguida de manifestaciones cerebelosas y de tronco, siendo menos frecuentes los síntomas extrapiramidales, la afasia y la disartria. El curso clínico frecuentemente es recurrente y remite tras iniciar el tratamiento25. Las imágenes de RM muestran múltiples lesiones hiperintensas en regiones subcorticales y periventriculares. No obstante, este hallazgo puede aparecer en sujetos de edad avanzada así como en otras entidades, como la hipertensión o la diabetes. En la evaluación con SPECT se describen áreas de hipoperfusión cortical frontoparietal. Otras pruebas complementarias que apoyan el diagnóstico en el episodio agudo son el electroencefalograma o el análisis de líquido cefalorraquídeo (LCR), el cual muestra una intensa pleocitosis y bandas oligoclonales en el estudio electroforético.
La afectación de la médula espinal no es infrecuente en el SSP. Puede ser aguda o crónica y asociarse con manifestaciones encefálicas. En la serie de Delalande et al.8 representa el 50% de la afectación del SNC. Se ha descrito un caso de síndrome de Brown Sequard asociado al SSP, así como casos de mielitis recidivante, mielitis progresiva y hemorragia subaracnoidea espinal. Las lesiones se localizan generalmente a nivel cervical y dorsal. Las imágenes de RM se corresponden en la mayoría de los casos con una mielitis transversa extensa, con afectación de más de una metámera. No obstante, el estudio radiológico puede ser normal, lo que enfatiza la relevancia del diagnóstico desde el punto de vista clínico y electrofisiológico.
Tanto las mielopatías crónicas como la afectación focal del SNC pueden simular una esclerosis múltiple (EM) de forma progresiva. La serie de Alexander et al.1 describe 20casos de cuadros similares a EM en pacientes que presentan una mielopatía crónica. Otros autores describen su frecuencia a la inversa, investigando a pacientes diagnosticados de EM que cumplen criterios diagnósticos de SSP. En este tipo de estudios los resultados son discordantes, y su prevalencia oscila entre el 0 y el 16%26-28. La presentación clínica es superponible a una EM. No obstante, en el SSP la edad de inicio es superior (mayor de 40años), así como la ratio mujeres/hombres. La presencia de anticuerpos anti-SSA/SSB favorece el diagnóstico de SSP. Por otra parte, los pacientes con EM presentan con mayor frecuencia bandas oligoclonales (hasta en el 90%), así como alteraciones específicas en las imágenes de RM. En el caso de pacientes con manifestaciones clínicas de un SSP y serología negativa, la presencia de anticuerpos anti-α-fodrina puede apoyar el diagnóstico, aunque su especificidad es baja.
Un subtipo característico en las neuropatías centrales focales es la neuropatía óptica retrobulbar (NOR). Puede presentarse como una pérdida súbita de visión bilateral. En la serie de Delalande et al.8 su frecuencia se cifra en el 16% de las manifestaciones neurológicas. Se han descrito casos de afectación asintomática, evidenciada mediante potenciales evocados. La NOR puede ser la manifestación inicial de un SSP en ausencia de un síndrome seco. Asimismo puede acompañarse de afectación medular, resultando en una neuromielitis óptica de Devic.
Para Alexander et al.1 las meningoencefalitis representan el 25% de las complicaciones neurológicas centrales difusas en el SSP. En los casos descritos, el inicio se encuentra siempre marcado por un síndrome meníngeo y confusional, a veces acompañado de mialgias. Su asociación con una vasculitis cerebral es discutible. El análisis del LCR muestra una meningitis linfocitaria aséptica con un recuento celular superior a 900células/mm3. Todos los pacientes muestran en la RM áreas puntiformes hiperintensas en regiones subcorticales y periventriculares. El curso clínico suele ser leve y recurrente, con tendencia a estabilizarse sin presentar secuelas.
La afectación de las funciones cognitivas superiores en el curso del SSP se ha descrito ampliamente. Los pacientes pueden presentar manifestaciones singulares, como un deterioro en la concentración o alteraciones en los exámenes psicométricos. Estos últimos incluyen una disfunción ejecutiva, alteraciones visuoespaciales y deficiencias en la memoria a corto y a largo plazo. La RM es normal hasta en el 80% de los casos o puede revelar lesiones en sustancia blanca en regiones subcorticales frontoparietales. Por otro lado, los pacientes pueden presentar una clínica más florida en forma de demencia. Se han descrito casos de demencia tipo Alzheimer, donde es de especial interés el uso de SPECT que muestra áreas de hipoperfusión en regiones frontales y temporales.
En la literatura médica se encuadran las manifestaciones psiquiátricas dentro de las manifestaciones encefálicas difusas. Según distintas series, representan hasta el 13-60%8,25 de las manifestaciones del SNC. El empleo de exámenes psicométricos, así como la presencia de alteraciones difusas en la RM, apoyan el diagnóstico orgánico en este tipo de manifestaciones clínicas. La mayoría de los casos descritos presentan depresión (30%) asociada a cansancio incapacitante (65%). Por otra parte, el SSP puede asociarse a un síndrome fibromiálgico en el 30% de los casos, sin objetivarse signos clínicos ni hallazgos de imagen que expliquen dichos síntomas.
TratamientoEl manejo de las manifestaciones neurológicas del SSP es empírico, ya que no existen ensayos clínicos controlados. El tratamiento óptimo —adaptado al estado de la cuestión— de las manifestaciones neurológicas del SSP se basa en la adaptación de la terapia a la afectación que presenta el paciente. En los casos en que se sospecha una afectación vasculítica del SNC o del SNP, o una patogenia predominante basada en el depósito de inmunocomplejos o en la secreción de autoanticuerpos neuronotóxicos, el tratamiento se basará en el uso de glucocorticoides a dosis altas e inmunosupresores como la ciclofosfamida o el micofenolato, siendo el rituximab una opción válida en casos refractarios (véase más adelante). Por otra parte, muchos tipos de neuropatía periférica predominantemente sensitiva responden escasamente a la inmunosupresión y el tratamiento sintomático se erige en la principal medida, pudiéndose en determinados casos ensayar las inmunoglobulinas intravenosas (IgIV) a dosis altas.
En el caso de neuropatía periférica de predominio sensitivo, es fundamental el control del dolor neuropático. Los pacientes con dolor quemante delimitado por un dermatoma o territorio de nervio periférico concreto presentan una sensibilización periférica y el control sintomático se basa en el uso de agentes antiepilépticos como la gabapentina o la pregabalina. En cambio, en los pacientes con sensibilización central, caracterizada por alodinia táctil y dolor no delimitado a un dermatoma o territorio de nervio periférico concreto, se recomienda el uso de agentes antidepresivos, como la duloxetina o la venlafaxina.
El tratamiento de las manifestaciones del SNC exige un abordaje terapéutico intensivo, ya que en la mayoría de los casos suponen una amenaza para la vida del paciente. Tanto en casos de mielopatía como en vasculitis del SNC se recomienda el uso de bolos de metilprednisolona seguidos de glucocorticoides a dosis altas más ciclofosfamida en pulsos mensuales intravenosos.
El uso de IgIV puede resultar beneficioso en pacientes con neuropatía sensorial y motora que no responden a glucocorticoides y otras terapias inmunosupresoras. No se conoce con certeza la dosis óptima de IgIV y no existen ensayos clínicos controlados. El típico curso de tratamiento es de 400mg/kg diarios durante 5días. Takahashi et al.29 trataron a 5pacientes con neuropatía atáxica sensorial refractaria a corticosteroides y agentes inmunosupresores con IgIV. Cuatro de ellos presentaron mejoría clínica. Su efecto fue de larga duración, sin aparente empeoramiento clínico.
Los linfocitos B juegan un papel crucial en el SSP, ya que actúan como célula presentadora de antígeno y producen tanto autoanticuerpos como citocinas30,31. Por ello, la terapia dirigida contra los linfocitosB es una atractiva alternativa terapéutica. El rituximab es un anticuerpo monoclonal (AcM) quimérico murino/humano anti-CD20, molécula expresada por diferentes tipos celulares del linaje ontogénico del linfocitoB pero no por las células plasmáticas. El tratamiento con rituximab produce una profunda depleción de linfocitosB periféricos. El rituximab se ha aprobado para su uso en artritis reumatoide, linfoma noHodking y leucemia linfática crónica. Los primeros estudios que evaluaron rituximab en el SSP fueron ensayos abiertos. Gottenberg et al.32 realizaron un estudio retrospectivo que incluyó a 6pacientes con SSP que asociaban linfoma o manifestaciones extraglandulares. Se obtuvieron mejorías significativas tanto en los síntomas subjetivos como en la estabilización en las pruebas diagnósticas y manifestaciones extraglandulares. Por otro lado, en el estudio retrospectivo de Seror et al.33 se observó una mejoría en las manifestaciones extraglandulares así como una disminución de los requerimientos de corticosteroides. Se han realizado 4ensayos clínicos aleatorizados y controlados con placebo de rituximab en el SSP. El primer ensayo clínico incluyó a 18pacientes que recibieron la pauta de rituximab que se emplea en la artritis reumatoide (2dosis de 1g separadas 2 semanas)34. A los 36meses de tratamiento solo se observó una mejoría significativa en la escala analógico-visual para el cansancio. No se obtuvo mejoría significativa en el test de Schirmer ni en los niveles de inmunoglobulinas y autoanticuerpos. El segundo ensayo clínico incluyó 30pacientes tratados también con la pauta de rituximab que se emplea en la artritis reumatoide35. A las 12semanas se observó una mejoría en la secreción salival en los pacientes tratados con rituximab así como en la escala analógico-visual para la sequedad oral. También se observó disminución en la frecuencia de manifestaciones extraglandulares. Para confirmar estos resultados prometedores, 2ensayos clínicos se encuentran actualmente activos. El primero de ellos36 es el estudio TEARS. Se trata de un estudio multicéntrico aleatorizado, dobleciego y controlado con placebo. Se han incluido 120pacientes que cumplen los criterios europeo-americanos para el SSP y que presentan enfermedad activa y la presencia de al menos una manifestación extraglandular. El segundo de ellos es el estudio TRACTISS37. Se trata de un estudio multicéntrico aleatorizado controlado con placebo, que incluirá a 100pacientes que cumplen los criterios europeo-americanos para el SSP y que presentan al menos una manifestación extraglandular. Recientemente se han utilizado los datos prospectivos del registro francés de autoinmunidad y rituximab para evaluar las indicaciones, la eficacia y la seguridad en pacientes con manifestaciones sistémicas en el SSP38. De los 78pacientes evaluados, en el 95% se usó el rituximab para tratar una afectación extraglandular refractaria, y en el 60% de los casos el clínico consideró eficaz el tratamiento. Meikinian et al.39 evaluaron la eficacia y la seguridad de rituximab en pacientes con SSP y afectación del SNC. Para ello utilizaron los datos prospectivos del registro francés de autoinmunidad y rituximab (AIR). De los 11pacientes incluidos en el estudio, no se observó mejoría clínica ni en las técnicas de imagen en los pacientes con cuadros similares a la EM ni en los que presentaban manifestaciones cognitivas. Solo se observó mejoría en un paciente con mielitis transversa y en otro con manifestaciones psiquiátricas. Ambos pacientes asociaron tratamiento con micofenolato mofetilo y ciclofosfamida, respectivamente. Los autores postulan como posibles causas del fallo terapéutico de rituximab a que se trataba de un grupo de pacientes refractarios a tratamientos previos y que presentaban una enfermedad de larga evolución. Son necesarios ensayos prospectivos que recluten un gran número de pacientes y puedan responder a cuestiones como resultados a largo plazo, riesgo de infección o desarrollo de cáncer y efectos adversos. Recientemente se han publicado casos de leucoencefalopatía multifocal progresiva (LMP)40 en pacientes tratados con agentes biológicos. El problema más temible es el riesgo de LMP, una enfermedad desmielinizante poco frecuente, progresiva y fatal causada por la reactivación del virus JC. Existe un riesgo acumulado de 2,2 casos de LMP por cada 100.000pacientes con artritis reumatoide tratados con rituximab30. No existen datos del riesgo asociado de LMP en pacientes con SSP, y se estima que puede ser similar al registrado en pacientes con artritis reumatoide. En todos los casos será necesaria la realización de un consentimiento informado en el que se notifique el potencial beneficio-riesgo de dicho tratamiento.
El epratuzumab es un AcM humanizado dirigido contra CD22, un antígeno de superficie específico de los linfocitos B. CD22 interviene en procesos de regulación de la señalización intracelular. Su uso no está aprobado en enfermedades autoinmunes y no existen ensayos clínicos en la actualidad para el SSP. Se han comunicado los resultados de un pequeño ensayo abierto en 16pacientes con SSP con afectación glandular41. El fármaco fue bien tolerado. Se realizó un índice compuesto por el test de Schirmer, flujo salival no estimulado, fatiga y VSG e IgG. Al menos se observó un 20% de mejoría en 2 de los parámetros que constituían una respuesta clínica. El fármaco produce una disminución de los linfocitosB circulantes, menos profunda que la evidenciada con rituximab, y no se ha observado una modificación significativa de los niveles de inmunoglobulinas. Epratuzumab podría ser un tratamiento prometedor para pacientes con SSP, siendo necesarios ensayos clínicos controlados por placebo.
ConclusionesLos pacientes con SSP pueden desarrollar diversas complicaciones extraglandulares que afectan al sistema nervioso central y periférico. Su adecuada evaluación permite en la mayoría de los casos establecer el régimen terapéutico óptimo, adaptado al tipo de afectación que presenta el paciente. El uso de dosis altas de glucocorticoides e inmunosupresores se reserva para los casos graves con compromiso vital o funcional relevante. En los casos refractarios, el uso de inmunoglobulinas intravenosas o de rituximab puede ser eficaz. Son necesarios ensayos clínicos controlados que definan la utilidad y la seguridad de los diferentes abordajes terapéuticos propuestos en base a la patogenia autoinmune-inflamatoria que se presume en las manifestaciones neurológicas del SSP.
Conflicto de interesesJ.L. Andreu ha recibido honorarios por conferencias de formación médica continuada de Roche y UCB y por asesorías científicas de UCB. El resto de los autores declara no tener conflictos de interés.