





El síndrome de resistencia a la insulina, actualmente más conocido como síndrome metabólico (SM), es una condición clínica de alta prevalencia en Chile. Por el mayor riesgo de enfermedades cardiovasculares y diabetes asociado a esta condición, en la práctica clínica es relevante su detección precoz y manejo. La resistencia a la insulina (RI) es una condición metabólica central en la etiopatogenia del SM y su diagnóstico puede efectuarse con mediciones de insulinemia y glicemia en ayuno o con la prueba de tolerancia oral a la glucosa con curva de insulinemia. Existe acuerdo que los mejores resultados del manejo del SM y de la RI se logran con cambios en estilo de vida, incluyendo modificaciones en la dieta y en el patrón de actividad física junto con reducción en el peso y grasa corporal. Algunas terapias farmacológicas orientadas a mejorar la sensibilidad a la insulina han sido recomendadas en consensos internacionales, especialmente en pacientes con elevado riesgo de enfermedad cardiovascular o de diabetes tipo 2, cuando fracasan las terapias no farmacológicas.
Insulin resistance syndrome, now known as metabolic syndrome (MS) is a highly prevalent clinical condition in Chile. Because of the high associated risk of this condition with cardiovascular disease and type 2 diabetes, in the clinical practice is relevant its early detection and management. Insulin resistance (IR) is a key metabolic condition in the pathogenesis of MS, and its diagnosis can be made with fasting plasma glucose and insulin measurements or by an oral glucose tolerance test with insulin curve. There is agreement that the best results of the management of MS and insulin resistance are achieved with lifestyle modifications, including changes in diet and physical activity patterns along with weight reduction and body fat. Some pharmacological therapies targeted on improving insulin sensitivity, have been recommended in international consensus, especially in patients at high risk of cardiovascular disease or type 2 diabetes, when nonpharmacological strategies have failed.
Los patrones dietarios y de actividad física han cambiado drásticamente en las últimas décadas, lo cual ha contribuido al rápido aumento de la prevalencia de obesidad y de sus múltiples complicaciones asociadas (1). Entre las más prevalentes se encuentran la resistencia insulínica (RI), dislipidemia e hipertensión arterial, las cuales conforman el así llamado síndrome metabólico (SM). En Chile existe una alta prevalencia de síndrome metabólico, comprometiendo a cerca de un tercio de la población adulta (2).
Diagnóstico del síndrome metabólicoEl SM fue conocido inicialmente como síndrome X, síndrome de resistencia a la insulina o síndrome de Reaven. Este síndrome se caracteriza por una disminución de la sensibilidad a la insulina, junto con obesidad (central), dislipidemia, hiperglicemia, hipertensión arterial, inflamación crónica y mayor propensión a la enfermedad trombótica (3). En 1998 la Organización Mundial de la Salud (OMS) propuso la denominación de SM para englobar las distintas entidades patológicas que determinan un aumento del riesgo cardiovascular (4). Estudios poblacionales prospectivos han mostrado que el SM aumenta alrededor de 2 veces el riesgo de eventos por enfermedad vascular aterosclerótica (5, 6) y además aumenta entre 3,5 y 5 veces el riesgo de diabetes tipo 2 (7).
Varias definiciones han sido propuestas por distintos grupos, entre ellos la OMS (1998 y 1999), el Grupo Europeo para el Estudio de la RI (EGIR-1999), el Programa Nacional de Educación del Colesterol de Estados Unidos (NCEP-ATP III 2001) y la Asociación Americana de Endocrinólogos Clínicos (AACE-2003) (8). En abril de 2005 la Federación Internacional de Diabetes (IDF) planteó una nueva definición del SM, aunque no se fundamentó cuáles fueron los nuevos hallazgos epidemiológicos o clínicos que justificaron la nueva definición. Este consenso redefinió los puntos de corte, utilizando los mismos parámetros del ATP III, y confiriendo a la obesidad abdominal un papel fundamental. Así, se definió como saludable una circunferencia de cintura inferior a 80 y 94cm, en mujeres y hombres, respectivamente (9). Estos valores de cintura fueron definidos por la OMS en 1997, basándose en el aumento del riesgo de enfermedades metabólicas observado en estudios que mostraban concordancia con un índice de masa corporal (IMC) de 25kg/m2 (10). La nueva definición de la IDF elevó la prevalencia de SM en forma importante en la mayoría de los países. Esta definición estableció que la obesidad abdominal era un requisito para el diagnóstico de SM, por lo que entre los sujetos portadores, una mayor proporción eran obesos. En cambio, el criterio de ATP III da importancia a todos sus factores por igual y como 2 de los 5 criterios están relacionados con los lípidos sanguíneos (colesterol de HDL y triglicéridos), detecta con mayor frecuencia a sujetos con dislipidemia (11). El año 2009, un consenso de varias organizaciones líderes acordó mantener los criterios de la ATP III-2005, considerando la presencia de SM con al menos 3 de 5 criterios, debiendo definirse en distintas regiones o países el punto de corte de cintura, dependiendo de las características particulares de cada población (12) (Tabla 1).
Criterios diagnósticos
| Factores de riesgo | IDF 2005 (Circ. cintura y ≥2 otros criterios) | AHA/NHLBI 2005 (≥3 criterios) | IDF/NHLBI/AHA/WHF/IAS/ISAO 2009 (≥3 criterios) |
|---|---|---|---|
| Circ. cintura (cm) | ≥94 en hombres (H); ≥80 en mujeres (M) | ≥102 (H); ≥88 (M) | Punto de corte a definir en cada región |
| Glucosa (mg/dL) | ≥100 ó diabetes tipo 2 | ≥100 | ≥100 |
| Presión arterial (mm/Hg) | ≥130 PAS ó ≥85 PAD* | ≥130 PAS ó ≥85 PAD | ≥130 PAS ó ≥85 PAD |
| Colesterol-HDL (mg/dL) | <40 (H); <50 (M)* | <40 (H); <50 (M) | <40 (H); <50 (M) |
| Triglicéridos (mg/dL) | ≥150* | ≥150 | ≥150 |
En la práctica clínica, identificar a un sujeto como portador de SM puede presentar dificultades técnicas, considerando que varios parámetros muestran una alta variabilidad intra-individual (ej. trigliceridemia, glicemia y presión arterial). Por otro lado, las recomendaciones para medir la circunferencia de cintura difieren entre distintos organismos, junto con ser dependiente de la experiencia del evaluador. De tal forma, por el carácter dicotómico con que se evalúa la presencia o no de cada criterio, un sujeto puede ser identificado como portador o no de SM (13).
Diagnosticar a un individuo como portador de SM es útil en clínica para pesquisar a aquellos individuos con mayor riesgo cardiovascular y de diabetes tipo 2 y de esa forma implementar en ellos medidas preventivas. Sin embargo, es discutible que con un manejo dicotómico de las variables (ej. glicemia menor o mayor a 100mg/dL) se pueda definir el nivel de riesgo cardiovascular o de diabetes tipo 2 a nivel individual. Esto dado que la relación entre estas variables y el riesgo de enfermedad es continuo. Respecto del riesgo de diabetes tipo 2, este estará incrementado con la sola presencia de un valor elevado de glicemia de ayuno, aún sin cumplir el paciente con los otros criterios mínimos de SM. En cambio, otros pacientes con SM, pero sin glicemia alterada de ayuno podrían tener menor riesgo de diabetes tipo 2. Adicionalmente, respecto de la relación entre SM y RI, distintos estudios muestran que no todos los pacientes con SM presentan RI, observándose un valor predictivo del SM para detectar esta condición metabólica entre 50 y 78% (13).
Definición de resistencia insulínicaLa insulina es una hormona anabólica secretada por las células â del páncreas en respuesta a diversos estímulos, siendo la glucosa el más relevante (14, 15). Su principal función es mantener la homeostasis glicémica y de otros sustratos energéticos. De esta forma, posterior a cada comida la insulina suprime la liberación de ácidos grasos libres mientras que favorece la síntesis de triglicéridos en el tejido adiposo. Por otra parte, la insulina inhibe la producción hepática de glucosa, mientras que promueve la captación de glucosa por el tejido muscular esquelético y adiposo (16). En un estado de RI, la acción de esta hormona a nivel celular está reducida, lo que aumenta la secreción de insulina. Esto permite compensar el defecto en la acción tisular y así mantener la homeostasis glicémica (17). Este fenómeno da cuenta del estado hiperinsulinémico, el cual es característico en sujetos con RI, particularmente después de una comida alta en carbohidratos.
Evaluación de la resistencia insulínicaLa RI puede ser determinada mediante un clamp euglicémico-hiperinsulinémico (18). Esta técnica consiste en infundir insulina a una tasa fija, mientras se administra glucosa a una tasa variable con el objeto de fijar (clamp) la glicemia a un nivel dado, usualmente 90mg/dL. En sujetos con menor grado de RI (sensibles a insulina) se requerirá una mayor tasa de infusión de glucosa para mantener la euglicemia. La aplicación de este método es compleja, laboriosa y costosa, lo cual ha incentivado el desarrollo de otras métodos para evaluar la RI fundamentalmente basadas en estimaciones de la glicemia e insulinemia en ayuno o en respuesta a una dosis oral estándar de glucosa (19).
Evaluación de la resistencia insulínica basada en mediciones de ayunoPor su simplicidad y buena correlación con mediciones más complejas de sensibilidad a la insulina, el método más utilizado es el cálculo del índice HOMA-IR (Homeostasis Model Assessment of Insulin Resistance) (20):
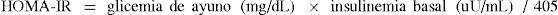
El punto de corte para definir resistencia insulínica de acuerdo a éste índice fue primero definido por Bonora et al (21) como el límite inferior del mayor quintil de HOMA-IR en 225 adultos con tolerancia normal a la glucosa e IMC < de 25kg/m2, pertenecientes al estudio Bruneck. Este valor correspondió a 2,77. En Chile, un estudio en 120 adultos aparentemente sanos entre 19 y 40 años, observó que el promedio más una desviación estándar correspondía a un índice HOMA de 2,5, proponiéndose así este valor como punto de corte para definir RI en la práctica clínica y para estudios poblaciones (22).
Otros investigadores han planteado que la sola medición de insulinemia basal puede ser un buen indicador de resistencia a la insulina, en sujetos con tolerancia normal a la glucosa, por su buena correlación con la sensibilidad a la insulina evaluada con el método de Clamp. En relación al punto de corte para definir RI, Laakso et al (23) observan que con un valor mayor de 13 uU/mL un 74% de los sujetos son resistentes a la insulina, y McAulley et al (24) consideran que un valor de 12,2 uU/mL presenta una buena relación entre sensibilidad y especificidad, aunque otros estudios han planteado puntos de corte más elevados de alrededor de 16 uU/mL (25, 26) pero en base a un criterio de riesgo metabólico o cardiovascular asociado. El punto de corte de 12uU/ml es el más utilizado en la práctica clínica, además de concordar con un índice HOMA de 2,5 cuando se considera el punto medio del rango normal de glicemia de ayuno (85mg/dL). En general se ha demostrado que los indicadores de RI basados en mediciones de ayuno representan la resistencia a la insulina hepática, y que no muestran una buena correlación con la RI periférica (muscular y/o de adipocitos), ni son buenos indicadores de RI en sujetos con glicemia alterada de ayuno o diabetes (23).
Resistencia a la insulina evaluada por curva de insulinemia y test de tolerancia oral a glucosaLa prueba de tolerancia oral con 75 gramos de glucosa que se utiliza en la práctica clínica para diagnosticar estados de intolerancia a glucosa o diabetes, al agregar mediciones de insulinemia en cada momento de medición de la glicemia, permite pesquisar la existencia de RI cuando se observan valores de insulinemia mayores de 100 y 60 uU/mL, a la hora y 2 horas de la carga de glucosa, respectivamente, con la condición de que la glicemia sea menor de 140mg/dL a las 2 horas de la prueba (ausencia de intolerancia a glucosa) (25, 26). Si bien estos puntos de corte para insulinemia son usados en la práctica clínica como elementos de sospecha de RI, no han sido validados como estándares universales para efectuar un diagnóstico preciso.
Un método de investigación que puede ser usado en clínica es el índice de Matsuda que requiere 5 mediciones de glicemia e insulinemia en la prueba de tolerancia oral a la glucosa, definiendo RI corporal total con un valor menor de 2,5 según el resultado de la siguiente fórmula (27):
Otros métodos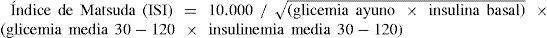
El test de tolerancia a la glucosa intravenosa con muestreo frecuente modificado (FSIVGTT), basado en el modelo mínimo de Bergman et al (28), es una prueba usada con fines de investigación para evaluar tanto la secreción de insulina como la sensibilidad a ésta. Es una prueba laboriosa que considera la extracción de muestras de sangre venosa en 30 ocasiones durante un periodo de 3 horas, además de la infusión endovenosa de glucosa y de insulina en dosis estandarizadas. Los datos se analizan mediante un programa computacional para calcular respuesta insulínica de primera fase, sensibilidad a la insulina, eficacia de la glucosa, e índice de disposición de glucosa (19).
También se ha utilizado la excreción urinaria de péptido C (molécula co-secretada con insulina) relativa a la ingesta alimentaria, como un indicador más fisiológico de la RI (29).
La determinación de la RI, cualquiera sea la forma de hacerlo (métodos de referencia o estimaciones), se realiza fundamentalmente con fines de investigación. La determinación de la RI con fines diagnósticos es de dudosa confiabilidad dada la alta variabilidad existente entre los métodos de determinación de la insulina (30), lo cual impide definir puntos de corte y en consecuencia clasificar de manera universal a los individuos según su grado de RI (31).
Factores patogénicos de la resistencia a la insulinaResistencia insulínica mediada por inflamaciónLa inflamación es uno de los mecanismos fisiopatológicos por los cuales se puede condicionar la RI. La obesidad ha sido asociada a un estado inflamatorio crónico leve a moderado, el que se manifiesta a nivel sistémico por un aumento de los factores inflamatorios y los leucocitos circulantes (32). A nivel tisular y particularmente en el tejido adiposo, se caracteriza por infiltración de células inmunes. A nivel molecular, diversos tipos celulares (adipocitos, células endoteliales, leucocitos, células hepáticas, célula â pancreática, neuronas, entre otras) manifiestan una mayor unión de factores de transcripción pro-inflamatorios (por ejemplo el factor nuclear kappa Beta o NFκB) a elementos de respuesta nuclear (33).
En condiciones pro-inflamatorias, los mediadores inflamatorios se unen a los receptores de las membranas celulares, lo cual desencadena la migración del factor de transcripción NFêB desde el citosol al núcleo para la síntesis de nuevos mediadores inflamatorios. En estado basal, este factor de transcripción está inactivo en el citosol, unido a su inhibidor IêB, lo que le impide migrar al núcleo. En respuesta a una señal externa pro-inflamatoria (ej. TNFá), la proteína IKK induce la degradación de IêB, dejando a NFêB libre para migrar al núcleo, transmitiendo así la señal inflamatoria. Sin embargo, la proteína IKK también fosforila al sustrato del receptor de insulina 1 (IRS1). En condiciones fisiológicas, IRS1 se activa cuando está fosforilado en residuos de tirosina; sin embargo, la fosforilación de IKK ocurre en su residuo serina. Como consecuencia, hay una inhibición de la transducción de la señal insulínica, determinando una menor translocación del transportador de glucosa 4 (GLUT4) desde el citosol a la membrana celular, disminuyendo así la captación de la glucosa sanguínea (Figura 1). Como respuesta compensatoria, ocurre una hipersecreción de insulina, lo cual explica la típica hiperinsulinemia de los individuos con RI (33, 34). De esta manera, una célula expuesta a un entorno inflamatorio es una célula resistente a insulina.
![Eventos celulares que interfieren en la señalización insulínica La insulina (triángulo) se une a su receptor de membrana, gatillando la fosforilación del mismo receptor y proteínas post-receptor (ej. sustrato del receptor de insulina 1 [IRS1], Akt, etc.). Esto determina la migración del transportador de glucosa 4 (GLUT4) a la membrana, lo que facilita la captación de glucosa. En un proceso inflamatorio, los mediadores inflamatorios se unen a su receptor de membrana activando (línea negra continua) a proteínas quinasas (ej. IKK y JNK). Estas proteínas inhiben a IRS1 (líneas punteadas) reduciendo la señalización insulínica. Asimismo, las especies reactivas del oxígeno (EROs) y el estrés del retículo endoplásmico (ER) estimulan a IKK y JNK. Lípidos específicos también interfieren en la señalización insulínica al activar la proteína kinasa C (por diglicéridos), la cual inhibe a IRS1, o reducir la actividad de Akt (por ceramidas).](https://static.elsevier.es/multimedia/07168640/0000002400000005/v4_201611160319/S071686401370230X/v4_201611160319/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd2Vt2E9KIXSbfPNY5VCUB4kpCjPTZRm5n9r2Wgu2xKnaaLdMEH3EdygauzKlTyPTxzYv7Zmk9BQK1ts1U3i/WIptqRpapvx4DrqsLcA/JvIwuIQQQY7TNmOVL4dGrUYtHw+Wc7rZgoebgUEMgtKOVrQN2pveRFWZoTJyywn7ZHroEf/ic4mIqsmFGyDuFZZxv4tqwxnCYNoex2C1Mq7H7/Vb0SCkmpk2yPOW2fZpIKpzokVGUYDMB5FTc9wEIc+gtVcoFEfrP5vayTpfFOf6Zi "Eventos celulares que interfieren en la señalización insulínica La insulina (triángulo) se une a su receptor de membrana, gatillando la fosforilación del mismo receptor y proteínas post-receptor (ej. sustrato del receptor de insulina 1 [IRS1], Akt, etc.). Esto determina la migración del transportador de glucosa 4 (GLUT4) a la membrana, lo que facilita la captación de glucosa. En un proceso inflamatorio, los mediadores inflamatorios se unen a su receptor de membrana activando (línea negra continua) a proteínas quinasas (ej. IKK y JNK). Estas proteínas inhiben a IRS1 (líneas punteadas) reduciendo la señalización insulínica. Asimismo, las especies reactivas del oxígeno (EROs) y el estrés del retículo endoplásmico (ER) estimulan a IKK y JNK. Lípidos específicos también interfieren en la señalización insulínica al activar la proteína kinasa C (por diglicéridos), la cual inhibe a IRS1, o reducir la actividad de Akt (por ceramidas).")
Eventos celulares que interfieren en la señalización insulínica
La insulina (triángulo) se une a su receptor de membrana, gatillando la fosforilación del mismo receptor y proteínas post-receptor (ej. sustrato del receptor de insulina 1 [IRS1], Akt, etc.). Esto determina la migración del transportador de glucosa 4 (GLUT4) a la membrana, lo que facilita la captación de glucosa. En un proceso inflamatorio, los mediadores inflamatorios se unen a su receptor de membrana activando (línea negra continua) a proteínas quinasas (ej. IKK y JNK). Estas proteínas inhiben a IRS1 (líneas punteadas) reduciendo la señalización insulínica. Asimismo, las especies reactivas del oxígeno (EROs) y el estrés del retículo endoplásmico (ER) estimulan a IKK y JNK. Lípidos específicos también interfieren en la señalización insulínica al activar la proteína kinasa C (por diglicéridos), la cual inhibe a IRS1, o reducir la actividad de Akt (por ceramidas).
La RI está comúnmente asociada a desórdenes del metabolismo lipídico que incluye la acumulación tisular ectópica de lípidos, entre ellos en el músculo esquelético (35, 36). Esta relación no es sólo asociativa, sino que existe evidencia concluyente, tanto en animales como en humanos, que los lípidos pueden inducir RI. En efecto, mediante una infusión endovenosa de una emulsión lipídica o una dieta hipercalórica e hipergrasa se induce RI en el lapso de algunas horas (infusión) a días (dieta) (37).
Existen dos aspectos que requieren ser discutidos para una mejor comprensión de la relación entre los lípidos y la RI. Por una parte, identificar cómo los lípidos se acumulan en tejidos ectópicos. Por otra parte, cómo y cuáles son las especies lipídicas que inducen RI. Sobre el primer punto, es claro que debe existir un desequilibrio entre la captación y oxidación de ácidos grasos que permita su acumulación en células de tejidos específicos. En general, la evidencia sugiere que la captación parece no estar elevada en sujetos con RI (38). Por otra parte, individuos con RI tienden a caracterizarse por una menor densidad mitocondrial y síntesis de ATP en músculo esquelético (39). Basado en esta evidencia, algunos autores han propuesto la existencia de una disfunción mitocondrial en músculo esquelético de individuos con RI, lo cual determinaría una menor capacidad oxidativa de ácidos grasos y, en consecuencia, su acumulación intracelular (38, 40).
Sin embargo, esta hipótesis ha sido ampliamente cuestionada, dado que una menor densidad mitocondrial no necesariamente determinará una menor oxidación de lípidos. En efecto, estudios en animales y humanos así lo indican (39). Por otra parte, la síntesis de ATP está fundamentalmente regulada por su demanda, por lo que una menor síntesis de ATP puede también ser interpretado como un estado de menor demanda energética muscular (39, 41). En definitiva, los determinantes de la acumulación ectópica de lípidos, particularmente, a nivel del músculo esquelético corresponden a un área de intenso estudio.
El segundo aspecto está referido a cómo los lípidos interfieren en la señal insulínica. Esto conduce a la pregunta de cuál u cuales especies lipídicas ejercen dicho efecto. Dado que los triglicéridos acumulados en músculo esquelético poseen una actividad biológica neutra, es decir, no interfieren en la actividad de proteínas, otras especies lipídicas debieran dar cuenta del efecto deletéreo sobre la señal insulínica. En este sentido, los diglicéridos o ceramidas han mostrado estar aumentados en músculo esquelético de sujetos con RI (42, 43).
Respecto a cómo los lípidos ejercen su acción inhibitoria sobre la señal insulínica, la evidencia disponible es más concluyente. Se ha demostrado que los diglicéridos son capaces de influenciar la actividad de proteínas específicas, entre ellas, la proteína quinasa Cè (41). Esta proteína posee actividad serín-quinasa, es decir, fosforila a proteínas blanco en sus residuos de serina. Uno de los sustratos para la acción de proteína quinasa Cè es IRS1, lo cual determina una atenuación de la actividad de la señal insulínica, de manera análoga a lo que ocurre en una condición proinflamatoria (Figura 1).
La RI mediada por lípidos también posee un nexo con la inflamación. En efecto, los ácidos grasos libres circulantes constituyen un estímulo pro-inflamatorio, dada su capacidad de unirse a receptores de membrana como TLRs (Toll-like receptors) (44). Estos receptores median la respuesta inmune innata. La activación de TLRs activa al NFêB, con la consiguiente liberación de citoquinas pro-inflamatorias. Estos receptores no son exclusivos de células inmunes, también se expresan en adipocitos, representando un nexo entre el exceso de nutrientes, en este caso lípidos, la inflamación y la inducción de la RI (45).
Tejido adiposo: órgano central en la inflamación y resistencia insulínicaEl principal tipo celular que compone el tejido adiposo (TA) es el adipocito, célula capaz de almacenar triglicéridos (TG) en su citoplasma sin ver afectada su fisiología. El tamaño de la gota lipídica del citoplasma está regulado por múltiples mecanismos, que en general incluyen la lipogénesis (formación de TG) y lipólisis (degradación de TG con salida de ácidos grasos libres a la circulación) (46). En condiciones de balance energético positivo crónico, esta célula puede expandir su volumen hasta 1000 veces. El adipocito hipertrófico tiene una mayor tasa lipolítica, lo cual condiciona una mayor liberación de ácidos grasos no esterificados a la circulación, por lo tanto, mayor riesgo de acumulación ectópica de lípidos. Por otra parte, los adipocitos de gran tamaño poseen una mayor síntesis y liberación de productos de secreción del tejido adiposo (adipoquinas) que pueden deteriorar el metabolismo lipídico y glucídico, tener efectos pro-inflamatorios o pro-trombóticos, además de inhibir la diferenciación de pre-adipocitos en adipocitos (47). Existen múltiples adipoquinas de efectos delétereos cuya secreción está aumentada en los adipocitos hipertróficos, entre las que destacan la leptina, resistina, angiotensina, citoquinas pro-inflamatorias y quemoquinas (48). Paralelamente, estos adipocitos de gran tamaño secretan menor cantidad de adiponectina, una de las pocas aipoquinas con efectos antagónicos a los recién descritos. Producto de este ambiente auto/paracrino pro-inflamatorio, el TA es infiltrado por macrófagos, que a su vez secretan moléculas pro-inflamatorias, alterando aún más el perfil secretor del TA, lo que perpetúa el fenómeno (49, 50). Lo descrito anteriormente sustenta la observación que el tamaño del adipocito del tejido adiposo abdominal subcutáneo se relaciona de manera directa con la RI, siendo también un factor de riesgo de diabetes tipo 2 (51, 52).
En contraposición con lo que se pensaba algunas décadas atrás, el número de adipocitos no es estático. En efecto, existen células precursoras en el estroma del tejido adiposo que bajo los estímulos adecuados pueden diferenciarse a células adiposas maduras (53). La hiperplasia, es decir, proliferación de precursores y posterior diferenciación a adipocitos, sería beneficiosa en condiciones de obesidad, pues disminuiría la necesidad de hipertrofiar las células adiposas y de esta forma prevenir el depósito ectópico de grasa (54).
Todo lo anterior permite plantear que las características de la expansión de la masa adiposa influirán de forma importante en el desarrollo de alteraciones metabólicas propias de la obesidad, entre ellas la RI. Aunque la evidencia es aún muy limitada, un estudio reciente indica que esto sería efectivo. En dicho estudio se señala que los obesos sin RI (medidos por clamp euglicémico-hiperinsulinémico) presentan un menor depósito adiposo visceral, una mayor cantidad de adipocitos pequeños, mayores niveles séricos de adiponectina, menor infiltración de macrófagos y células adiposas de mayor sensibilidad a insulina que las procedentes de sujetos obesos con RI (55).
De esta forma, un TA que se expande sin alterar sus características biológicas, podría condicionar una obesidad inocua desde el punto de vista metabólico y cardiovascular. Es así que una alternativa para prevenir las complicaciones asociadas a la obesidad sería favorecer la expansión de la masa adiposa (56). Existen diversos modelos en ratones que demuestran esta hipótesis (57, 58). En tanto, en humanos este mecanismo protector podría estar operando en respuesta al tratamiento con agentes farmacológicos como las tiazolidinedionas (agonistas de PPARã2). En efecto, estos pacientes usualmente manifiestan un aumento de la masa adiposa concomitante con una reducción de la RI.
Manejo nutricional del síndrome metabólico con énfasis en resistencia insulínicaEl papel de la dieta en la determinación del SM ha sido ampliamente estudiado. Sin embargo, dada la complejidad de la dieta, particularmente en humanos, la identificación de un patrón dietario específico, y más aún, de componentes nutricionales que modifiquen el riesgo de padecer de SM ha sido una tarea difícil. Por ejemplo, el estudio de los lípidos dietarios puede contemplar aspectos relacionados con la cantidad de grasa ingerida, como también el contenido de tipos de ácidos grasos en relación con el grado de insaturación, longitud de cadena, isomería geométrica, posición de los dobles enlaces, etc. Por otra parte, aislar una variable dietética en particular con el fin de identificar su influencia en el SM requiere de períodos de exposición prolongados, los cuales son a menudo difíciles de controlar en humanos. En esta sección haremos una síntesis de los principales componentes nutricionales y su relación con el SM, en especial con la RI.
El exceso de masa corporal es uno de los principales determinantes del grado de RI del individuo (35). De esta forma, la restricción energética asociada a pérdida de masa corporal es la principal intervención nutricional indicada en el tratamiento del SM (59). La pérdida de masa corporal y en especial de grasa corporal corrige múltiples anormalidades metabólicas, entre ellas, disminuye la RI, dislipidemia, hipertensión arterial, inflamación, etc. En general, la intervención orientada a limitar la ingesta excesiva de grasa, aumentar el consumo de frutas y verduras, y realizar más ejercicio físico es recomendable. En este sentido, Lindström et al. (60) observó una reducción en la proporción de sujetos que progresaron de intolerancia a la glucosa a franca diabetes tipo 2 después de aplicar estas guías.
Un aspecto estudiado ha sido el impacto de la composición nutricional de la dieta hipocalórica, en términos de la proporción de grasa y carbohidratos, sobre el control metabólico del individuo con SM. Kirk et al. (61) suministró dos tipos de dietas hipocalóricas (~1100 kcal/día por 11 semanas) en individuos con obesidad. Una de ellas con alto contenido de carbohidratos y bajo de grasa (65% y 20%, respectivamente), mientras que la otra tuvo un bajo contenido de carbohidratos y alto de grasa (10% y 75%, respectivamente). En ambos casos se observó una pérdida similar de masa y grasa corporal, como también una disminución similar del grado de RI. En la misma línea, un estudio multicéntrico Europeo observó después de 10 semanas de consumir una dieta hipocalórica baja (25% de la energía) y alta (40% de la energía) en grasa, una reducción comparable de la masa corporal, glicemia e insulinemia (62).
Ante condiciones isoenergéticas, es decir, de balance energético nulo, se ha evaluado el efecto del reemplazo de carbohidratos por grasa dietaria. En los estudios realizados, los cuales incluyen variaciones de la proporción de grasa dietaria entre 15% a 83% de la energía total y en los cuales la medición de RI ha sido en su mayoría realizada mediante métodos de referencia, se observó que la variación de la razón grasa a carbohidratos posee una modesta o nula influencia sobre el grado de RI (63).
La composición de ácidos grasos dietarios es otro componente que reviste interés nutricional. En animales, los ácidos grasos saturados, como aquellos derivados de oleaginosas, con alto contenido de ácido linoleico (ej. maravilla, girasol, etc.) han mostrado inducir RI. Por el contrario, ácidos grasos poliinsaturados de la serie n-3, particularmente aquellos encontrados en aceite de pescado, reducen la RI (64).
En tanto, en humanos los resultados son menos concluyentes en función del tipo de ácido graso dietario (64). Por una parte, es difícil garantizar la adherencia al régimen dietario en condiciones de vida libre. Por otra parte, las dosis usadas en humanos son varias veces inferiores a las utilizadas en animales. Aun así, un estudio que tuvo un adecuado diseño experimental en términos de duración de la suplementación, número de individuos estudiados, método de medición de la RI, entre otros, comparó la suplementación con aceite de oliva contra grasa saturada durante 3 meses (65). Se observó un leve aunque significativo aumento del grado de RI con la dieta suplementada en grasa saturada. Además, cuando se dividieron los individuos en función de la mediana de la ingesta de grasa, se observó que este efecto diferencial en el tipo de ácido graso era particularmente notorio en individuos con menor ingesta de grasa (<37% de la energía total). De esta forma, la recomendación nutricional tendiente a favorecer la ingesta de ácidos grasos monoinsaturados por sobre los saturados resulta aconsejable, en este caso, en términos de su impacto sobre la RI.
Los carbohidratos son un componente dietario mayoritario, los cuales tienen el mayor impacto en la glicemia. Por más de 30 años es sabido que los carbohidratos no afectan la glicemia de manera similar, lo cual dio origen al concepto de índice glicémico. Dado lo anterior, se ha planteado que el tipo de carbohidrato puede ejercer un efecto diferencial en el control metabólico del individuo (66).
Sin embargo, aislar el efecto nutricional de esta variable no es simple, dado que dietas de bajo vs. alta respuesta glicémica tienden a variar en contenido de fibra, antioxidantes, entre otros, lo cual impide ser concluyentes respecto al impacto específico del índice glicémico. Un metaanálisis de 18 estudios controlados mostró que dietas de bajo índice glicémico (incluía alto contenido de fibra y carbohidratos no disponibles) se asociaron con una reducción general del grado de RI de 20% (67). De manera específica, cuando se evaluó el efecto del índice glicémico en condiciones de balance energético nulo y ante dietas de contenido similar de carbohidratos, grasa y fibra, también se observó una reducción de la RI (68). Por lo tanto, parece aconsejable restringir el consumo de alimentos de alto índice glicémico, en especial en individuos con SM.
Un carbohidrato que ha despertado el interés dado su creciente disponibilidad dietaria y particular metabolismo hepático es la fructosa. Este carbohidrato posee un bajo índice glicémico, aunque no atribuido a una menor tasa de absorción intestinal, sino más bien a que la mayor parte de su metabolismo ocurre en el hígado y sólo una pequeña fracción se transforma en glucosa disponible para ser liberada a la circulación (69). La creciente disponibilidad de jarabe de maíz rico en fructosa ha estimulado el estudio del impacto de este carbohidrato en el SM. Los estudios que han comparado la ingesta de altas dosis de fructosa (>100g/dl) con glucosa han observado un aumento de la RI junto con mayor acumulación de grasa visceral y alteraciones del perfil lipídico (70). No obstante, es importante destacar que en EEUU más del 95% de la población adulta tiene una ingesta de fructosa menor a 100g/dl, por lo que los resultados obtenidos frente a dosis altas de fructosa pueden no ser aplicables en la mayoría de la población (71). Cuando se ha evaluado el efecto de dosis pequeñas de fructosa (10g por comida, < 30g/d) sobre parámetros metabólicos (ej. hemoglobina glicosilada, glicemia e insulinemia de ayuno) se observa una mejoría en el control metabólico del individuo (72). En este sentido, Hawkins et al. (73) encontró que la fructosa posee un efecto catalítico de la utilización hepática de glucosa, lo cual determina una menor producción de glucosa. Aunque no existe una recomendación especial que promueva la ingesta de fructosa, al menos el consumo de frutas, las cuales contienen fructosa, no está contraindicado en individuos con SM.
Otro componente importante de la dieta es la fibra y los carbohidratos no digeridos en el intestino delgado. Estas sustancias dietarias pasan al intestino grueso donde son fermentados por la flora residente. Producto de este proceso se liberan ácidos grasos de cadena corta, los cuales son un sustrato energético para la mucosa intestinal. Por otra parte, estos ácidos grasos pueden alcanzar otros órganos e impactar la sensibilidad insulínica a nivel sistémico (74). Esta información es sustentada por diversos estudios epidemiológicos que describen una relación inversa entre la ingesta de fibra y la prevalencia de SM y RI (75).
Un último aspecto a tratar en esta sección es la influencia de los antioxidantes sobre el control de la homeostasis glicémica y sensibilidad insulínica. El estrés oxidativo juega un papel importante en el control de la sensibilidad insulínica. Por una parte, las especies reactivas del oxígeno pueden inducir RI (Figura 1) (76). Por otra parte, se requiere un nivel mínimo (fisiológico) de estrés oxidativo para promover una adecuada transducción de la señal insulínica (77). Dado que la obesidad y el SM se caracterizan por un estado inflamatorio y mayor estrés oxidativo, mantener un adecuado suministro de sustancias antioxidantes parece ser una estrategia razonable. Además, el consumo de dietas pobres en frutas y verduras frescas, fortalece la idea de estudiar el impacto de una mejor capacidad antioxidante sobre el grado de RI.
En este sentido, Stull et al. (78), evaluaron mediante un ensayo clínico placebo-controlado, doble-ciego y aleatorio, el efecto de la suplementación por 6 semanas con un concentrado de arándanos en sujetos obesos con RI (medida mediante clamp euglicémico-hiperinsulinémico). Posterior a este período, la bebida rica en antioxidantes se acompañó de una reducción de la RI en comparación con la bebida placebo, sustentando el efecto favorable de antioxidantes naturales.
Un aspecto a considerar es el papel fisiológico que desempeña el estrés oxidativo en el control metabólico, por lo que el uso exagerado de antioxidantes pudiera tener consecuencias contrarias a la esperada. En efecto, el uso en humanos de megadosis de vitamina C (11 veces la recomendación) y E (27 veces la recomendación), provocó una disminución de la mejoría esperada en la RI después de 4 semanas de entrenamiento físico (79).
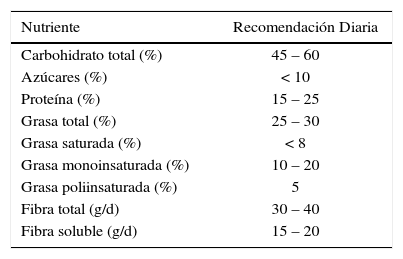
Un resumen de las recomendaciones nutricionales para el control del SM se presentan en la Tabla 2. Desde el punto de vista dietético, estas recomendaciones nutricionales pueden ser alcanzadas a través de una dieta balanceada, siendo la dieta mediterránea una alternativa. Esta se caracteriza por un adecuado aporte de frutas, vegetales, cereales, legumbres, pescados, frutos secos, vino tinto y aceite de oliva, y que en general puede englobar los distintos aspectos nutricionales descritos anteriormente, pudiendo ser altamente beneficiosa para el tratamiento del SM. En este sentido, una revisión de la literatura que incluyó 6 estudios prospectivos, mostró de manera global una reducción en el riesgo de cardiopatía coronaria (80).
Recomendaciones nutricionales para el control sm
| Nutriente | Recomendación Diaria |
|---|---|
| Carbohidrato total (%) | 45 – 60 |
| Azúcares (%) | < 10 |
| Proteína (%) | 15 – 25 |
| Grasa total (%) | 25 – 30 |
| Grasa saturada (%) | < 8 |
| Grasa monoinsaturada (%) | 10 – 20 |
| Grasa poliinsaturada (%) | 5 |
| Fibra total (g/d) | 30 – 40 |
| Fibra soluble (g/d) | 15 – 20 |
Actualmente se reconoce que las intervenciones basadas en cambios en los estilos de vida son la primera opción para el manejo del SM, aquellas que han sido exitosas revisten un alto grado de complejidad, como las aplicadas en el Programa de Prevención de Diabetes (81) y en el Estudio de Prevención de Diabetes de Finlandia (82). Esto condiciona un alto costo y un desafío para lograr una adecuada adherencia de los pacientes durante meses o años. Por otra parte, experiencias con intervenciones en los estilos de vida de menor costo y menos intensivas, también han mostrado revertir el SM y reducir la incidencia de diabetes tipo 2 (83).
Manejo farmacológico del síndrome metabólicoHasta el momento ningún fármaco ha mostrado la capacidad de reducir todos los factores de riesgo del SM, fundamentalmente porque la fisiopatología de esta condición es multifactorial (84). Alternativas farmacológicas que bloqueen el sistema renina–angiotensina, podrían potencialmente reducir la inflamación vascular, además de su efecto reductor de la presión arterial (85). Sin embargo, existe consenso en que el tratamiento farmacológico del SM debe restringirse al manejo de cada factor de riesgo de manera individual de acuerdo a las guías establecidas. Esto mientras no se disponga de estudios clínicos controlados que demuestren que un fármaco en particular posee la capacidad de actuar de modo multifactorial.
Una revisión sistemática y meta-análisis de 16 estudios (86), involucrando a 3907 pacientes con SM, mostró que la posibilidad de revertir el síndrome cuando se aplican estrategias para modificar los estilos de vida es 4 veces mayor que en los pacientes sin este tratamiento (odds ratio = 3,8; IC 95%: 2,5-5,9). En cambio, cuando se utilizan terapias farmacológicas (incluyendo hipolipemiantes, anti-diabéticos orales y fármacos anti-obesidad), la posibilidad de revertir el SM solo es un 60% mayor que en sujetos sin tratamiento (OR=1,6; IC 95%: 1,0-2,5). Al analizar las distintas combinaciones de tratamientos, los investigadores concluyeron que las mayores posibilidades de revertir el SM se observan con intervenciones de dieta más ejercicio, seguido por el uso de fármacos anti-obesidad (sibutramina o rimonabant) junto con consejos sobre estilos de vida y en tercer lugar dieta sola.
Manejo farmacológico de la resistencia a la insulinaConsiderando que la resistencia insulínica, es una condición metabólica que condiciona un mayor riesgo cardiovascular y de diabetes tipo 2, es cada vez más frecuente observar en la práctica clínica el uso de fármacos insulinosensibilizadores en sujetos sin diabetes tipo 2. Entre estos fármacos, el más usado es la metformina, aunque muchos estudios clínicos también han probado la eficacia de las tiazolidinedionas (rosiglitazona, pioglitazona) para tratar estados de resistencia a la insulina y de esta forma disminuir sus efectos deletéreos a largo plazo.
Las condiciones clínicas en que se ha estudiado con mejores resultados el manejo farmacológico de la resistencia a la insulina, especialmente con metformina, son la glicemia alterada de ayuno y/o intolerancia a la glucosa, el síndrome de ovarios poliquísticos e infertilidad asociada, la esteatosis hepática no alcohólica, y el síndrome metabólico y lipodistrofia asociado al tratamiento del VIH.
En estudios de prevención de diabetes se ha observado que las terapias farmacológicas que reducen la resistencia a la insulina disminuyen significativamente el riesgo de diabetes tipo 2, aunque cuando se comparan con terapia basada en cambios en estilos de vida, su efectividad no siempre es mayor. En el estudio DPP, el uso de metformina 850mg/2 veces al día redujo el riesgo de diabetes en 31% en comparación con un 58% logrado con cambios en estilos de vida (81). En el estudio de prevención de diabetes de India (87), la reducción del riesgo de diabetes después de 30 meses de seguimiento (28%), fue similar entre el grupo de estilo de vida y el grupo con metformina 1000mg al día, en cambio en un estudio efectuado en China (88), la rama de tratamiento con metformina 750mg al día logró una reducción significativamente mayor en el riesgo de diabetes que el grupo con cambios en estilo de vida (77% vs. 43%). Aún con esta evidencia, metformina no está indicada para la prevención de la diabetes en la mayoría de los países. Sin embargo consensos independientes de la American Diabetes Association (ADA) (89), y de la International Diabetes Federation (IDF) (90), han recomendado el uso de metformina, en conjunto con intervención sobre estilo de vida, en pacientes con glicemia alterada de ayuno o intolerancia a la glucosa, con edad menor de 60 años, antecedentes familiares de diabetes y presencia de obesidad u otros componentes del síndrome metabólico (hipertensión, HDL bajo, aumento de triglicéridos). La recomendación más reciente de la ADA establece que la indicación de metformina para la prevención de diabetes tipo 2 puede ser considerada en sujetos de mayor riesgo, como aquellos con múltiples factores de riesgo, especialmente si estos muestran progresión de la hiperglicemia (ej. HbA1C ≥6%), a pesar de intervención en estilo de vida (91). Otros fármacos sensibilizadores a la insulina, como las tiazolidinedionas, no se han considerado en las recomendaciones principalmente por su alto costo, perfil adverso de seguridad y falta de persistencia de su efecto a largo plazo.
Otra de las situaciones clínicas asociadas a resistencia insulínica en que se ha estudiado extensamente la aplicación de terapia farmacológica es el síndrome de ovario poliquístico (SOP) e infertilidad asociada. En estas pacientes la hiperinsulinemia se asocia a un aumento en el riesgo cardiovascular y de desarrollo de diabetes tipo 2 y los fármacos que mejoran la sensibilidad insulínica, tales como la metformina, se indican en la práctica clínica por su efectividad para tratar las manifestaciones clínicas del SOP, incluyendo la anovulación. Sin embargo, un reciente meta-análisis de 38 estudios que involucra a 3495 mujeres, concluye que metformina no mejora la tasa de nacidos vivos a pesar de aumentar la tasa de embarazos, ya se use sola o en combinación con clomifeno (92). Aún así, metformina ha demostrado ser de utilidad en mujeres con SOP para mejorar la composición corporal, disminuir factores de riesgo cardiovascular y las alteraciones hormonales y metabólicas que se asocian a este síndrome (93, 94).
La esteatosis hepática no alcohólica (EHNA), se considera en la actualidad como una manifestación más del síndrome metabólico, ya la que los pacientes con EHNA presentan con alta frecuencia resistencia a la insulina, obesidad, alteraciones del metabolismo de la glucosa, dislipidemia aterogénica e hipertensión arterial, aumentando el riesgo cardiovascular y contribuyendo a la progresión del daño hepático (95). Numerosos estudios han investigado el impacto de metformina en pacientes con EHNA, ya sea evaluando cambios en las enzimas hepáticas, en componentes del síndrome metabólico o en la histología (96). Estos estudios permiten concluir que aunque la terapia de elección para tratar la EHNA es la reducción de peso con cambios en estilo de vida, la metformina puede ser beneficiosa como terapia adjunta en pacientes con síndrome metabólico (97).
Los autores declaran no tener conflictos de interés, relacionados a este artículo.