








La osteogenesis imperfecta (OI) es un grupo de trastornos del tejido conectivo que genera anomalías esqueléticas caracterizadas por fragilidad y deformidades óseas. Las características genéticas son variables y se han descrito nuevos subgrupos los últimos años agregando información a las clasificaciones tradicionales. Su incidencia es de 1/10.000 a 20.000 RN vivos. Existe un amplio espectro de manifestaciones clínicas, que van desde una leve fragilidad ósea, en niños asintomáticos, hasta versiones que son letales al momento de nacer. El diagnóstico es principalmente clínico y debe diferenciarse de otras anomalías del esqueleto que producen fragilidad y de lesiones por maltrato infantil. El tratamiento es multidisciplinario y está orientado a mejorar la calidad de vida de los pacientes. Para lo que se debe mejorar la densidad ósea, a través de medicamentos, buena musculatura y cargas fisiológicas. Las fracturas se tratan con períodos cortos de inmovilización y carga precoz, o con cirugías que limiten el tiempo de inmovilización. Por otro lado, las deformidades esqueléticas deben tratarse en forma quirúrgica utilizando osteosíntesis que sean extensibles y mantengan la corrección a medida que el niño crece. El manejo coordinado de los distintos profesionales involucrados es de gran importancia para lograr los mejores resultados en esta enfermedad crónica que involucra al niño y todo su entorno.
Osteogenesis Imperfecta (OI) is a group of connective tissue disorders involved in skeletal abnormalities characterized by bone fragility and deformities. Genetic abnormalities are variable and new subgroups have been described recently, adding information to traditional classifications. There is a wide spectrum of clinical manifestations, ranging from mild bone fragility, in otherwise asymptomatic children, to versions that are lethal at birth. Its incidence is 1/10.000-20.000 newborns. The diagnosis is mainly clinical and must be distinguished from other skeletal abnormalities and child abuse. The treatment is multidisciplinary, and it is aimed to improve the quality of life of patients. For which the bone density must be improved, through medications, strong musculature, and physiological loads. Fractures are treated by immobilizing for short periods, trying to load at soon as possible, or by surgeries that limit immobilization time. On the other hand, skeletal deformities should be treated surgically using dynamic rods that are extensible and maintain correction as the child grows. The coordinated management of the different professionals involved is of the utmost importance to achieve the best results in this chronic disease that involves the child and his entire environment.
La fragilidad ósea, característica de la OI se produce en la mayoría de los casos por mutaciones estructurales o cuantitativas de los genes del colágeno 1 (COL1A1 y COL1A2). La primera descripción de una enfermedad hereditaria con fragilidad ósea fue hecha en una tesis doctoral de Ekman en 1788. Desde esa fecha la OI ha recibido numerosas denominaciones y sólo en el siglo XX se logró formar una unidad diagnóstica en aquellos pacientes que presentan como característica, fragilidad ósea, escleras azules y sordera.
La debilidad ósea está causada por: disminución de la masa ósea, alteración en la organización del tejido óseo y alteración en la geometría ósea (Figura 1).

Estudios histológicos han demostrado un aumento del recambio óseo en OI. Esto es lo que justifica el uso de bifosfonatos para reducir la reabsorción ósea mediada por osteoclastos, cuyos resultados han sido alentadores.
La administración cíclica de bifosfonatos reduce el dolor óseo y la incidencia de fracturas, aumentando la densidad ósea y el nivel de deambulación, con mínimos efectos colaterales. Su efecto en el hueso incluye aumento del tamaño de los cuerpos vertebrales y el engrosamiento del hueso cortical.
Estos resultados permiten una corrección quirúrgica más efectiva a través de enclavado endomedular e instrumentación paravertebral.
Programas específicos de fisioterapia y terapia ocupacional son muy importantes en el tratamiento de esta patología.
2ClasificaciónSe han descrito distintos tipos y clasificaciones, siendo la más clásica la de Sillence en 19791, quien describió 4 tipos, de acuerdo a la base genética y a las características clínicas, la que ha ido complementándose con nuevos subtipos, a medida que se conocen características moleculares específicas.
La mayoría de los tipos presentan alteraciones de uno de los dos genes que codifican el colágeno de tipo 1. En las formas recientemente descritas, se han encontrado defectos en otras proteínas (Síndromes de Bruck y Osteoporosis- Pseudoglioma).
2.1Clasificación de SillenceTipo I: herencia autosómica dominante. Corresponde al tipo más frecuente (dos tercios). Se caracteriza por osteoporosis generalizada, escleras azules por toda la vida y sordera de conducción. Es de intensidad leve, por lo que las fracturas son ocasionales y las deformidades escasas. De acuerdo a la existencia o no de dentinogénesis imperfecta se subdivide en A y B.
Tipo II: es la de mayor letalidad. Antiguamente se creía que tenía herencia autosómica recesiva debido a que la enfermedad se presentaba en hijos de padres sanos. Sin embargo, se demostró que la causa de esto era por mosaicismo del DNA de uno de los padres.
Presenta una elevadísima mortalidad perinatal debido a la extrema fragilidad ósea. Presentan fracturas a nivel de cráneo y tórax in útero y durante el parto, que acarrean diversas complicaciones letales.
Tipo III: herencia autosómica dominante. Presenta severa fragilidad ósea, con múltiples fracturas y numerosas deformidades. Las escleras son azulosas al nacer y se van haciendo más blancas hacia la adolescencia. Una imagen característica de este tipo es la epífisis en “pop corn”.
Tipo IV: herencia autosómica dominante. La severidad es algo mayor que la tipo I y con escleras blancas. La osteoporosis y fragilidad ósea es variable. De acuerdo a la dentición se clasifican en A y B.
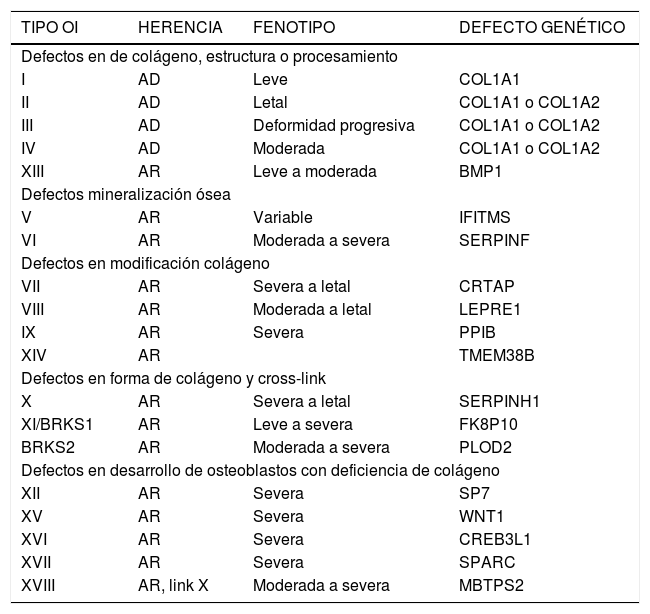
Muchos casos de osteogenesis imperfecta no se encuadran dentro de las cuatro categorías anteriormente descritas. Por ello se han agregado tres tipos más en los cuales no se han encontrado mutaciones en los genes para el colágeno I: tipo V, VI y VII2.
Tipo V: herencia autosómica dominante. Se caracterizan por desarrollar callos hipertróficos luego de una fractura, calcificación temprana de la membrana interósea en el antebrazo y bandas metafisiarias hiperdensas.
No presentan escleras azules ni dentinogénesis imperfecta.
La histología muestra que el hueso laminillar se agrupa de una manera irregular.
Tipo VI: se caracterizan por un defecto de mineralización. Tienen fracturas frecuentes, compresión vertebral y deformidad de huesos largos. Presentan densidad mineral ósea disminuída. Histológicamente hay abundante osteoide con ausencia de hipocalcemia, hipofosfatemia, o hipovitaminosis D.
Tipo VII:descrita en población nativa del norte de Quebec. Presentan acortamiento rizomélico de extremidades y coxa vara Tabla 1.
Clasificación expandida
| TIPO OI | HERENCIA | FENOTIPO | DEFECTO GENÉTICO |
|---|---|---|---|
| Defectos en de colágeno, estructura o procesamiento | |||
| I | AD | Leve | COL1A1 |
| II | AD | Letal | COL1A1 o COL1A2 |
| III | AD | Deformidad progresiva | COL1A1 o COL1A2 |
| IV | AD | Moderada | COL1A1 o COL1A2 |
| XIII | AR | Leve a moderada | BMP1 |
| Defectos mineralización ósea | |||
| V | AR | Variable | IFITMS |
| VI | AR | Moderada a severa | SERPINF |
| Defectos en modificación colágeno | |||
| VII | AR | Severa a letal | CRTAP |
| VIII | AR | Moderada a letal | LEPRE1 |
| IX | AR | Severa | PPIB |
| XIV | AR | TMEM38B | |
| Defectos en forma de colágeno y cross-link | |||
| X | AR | Severa a letal | SERPINH1 |
| XI/BRKS1 | AR | Leve a severa | FK8P10 |
| BRKS2 | AR | Moderada a severa | PLOD2 |
| Defectos en desarrollo de osteoblastos con deficiencia de colágeno | |||
| XII | AR | Severa | SP7 |
| XV | AR | Severa | WNT1 |
| XVI | AR | Severa | CREB3L1 |
| XVII | AR | Severa | SPARC |
| XVIII | AR, link X | Moderada a severa | MBTPS2 |
Abreviaciones: AD: autosómica dominante; AR autosómica recesiva
Es similar al tipo IV, pero es de herencia autosómica recesiva.
Se han descrito otros sindromes con fenotipos similares a OI en los cuales el defecto molecular no está en el colágeno:
- -
Síndrome de Bruck: contractura congénita de rodillas, tobillos y pies; en que hay una deficiencia de la telopeptido lisil hidroxilasa específica de hueso, que causa una aberrancia en el cross link del colágeno.
- -
Sindrome Osteoporosis Pseudoglioma: disminución de masa ósea, fracturas, deformidad y alteraciones oculares.
En 2015 fue descrita y luego complementada el 2017, la clasificación expandida basada en los defectos genéticos3–5,17.
2.3Clasificación clínicaEl 2014 se decribió una clasificación clínica, según fenotipo, más simple de recordar, que los agrupa en 5 grupos. (Tabla 2)4.
Clasificación clínica
| Nueva clasificación | Severidad, fenotipo | Tipo OI |
|---|---|---|
| 1 | Leve, no deformante | I |
| 2 | Severa, letal | II |
| 3 | Moderada a severa, deformidad progresiva | III, VI, VIII, IX, X, Sd. Bruck tipo 1 |
| 4 | Moderada | IV, VII, XI, XII, XIII |
| 5 | Moderada, incluye sindromes con calcificación de membranas interóseas | V, Sd. Osteoporosis-pseudoglioma, Osteoporosis idiopática juvenil, Sd. Bruck tipo 1 y 2 |
Antiguamente se consideraba que la OI tenía su origen en alteraciones exclusivamente del colágeno tipo 1. Con los avances en biología molecular y análisis genético, se ha comprobado la presencia de alteraciones en otras proteínas que generan síndromes muy similares de OI.
En las OI tipo I hasta la IV, la base de la enfermedad es una falla en la producción y organización del colágeno, debido a distintas mutaciones de los genes del colágeno ubicados en el brazo largo del cromosoma 17 y 7 (COL1A1 y COL1A2 respectivamente) y en errores de transcripción de éste. Han sido identificados más de 150 mutaciones específicas que pueden dañar estos genes y conducir a la OI.
La actividad de los osteoblastos es normal, pero la producción específica del colágeno tipo I se encuentra alterada, en cantidad o calidad. En la OI tipo I la alteración es en la cantidad de procolágeno sintetizada, siendo de aproximadamente la mitad que la de una célula normal. En los otros tipos de OI (II, III y IV) la alteración no es en la cantidad sino en la calidad del procolágeno sintetizado, lo que alteraría la estabilidad de las moléculas, impidiendo la formación normal de fibrillas de colágeno, siendo éstas delgadas y de distintos largos. Los proteoglicanos son normales.
El tejido óseo mantiene las proporciones de los componentes minerales y orgánicos, pero pierde la organización trabecular habitual. Los osteoblastos se encuentran en zonas de amplio material osteoide y aumentados en número. Grandes y numerosos osteocitos se encuentran rodeados de escasa cantidad de matriz. Los osteoclastos son numerosos y poseen una alta superficie de absorción ósea. Todos estos cambios son indicadores de un alto recambio óseo.
4Presentación clínica:El diagnóstico de la enfermedad es clínico y se caracteriza por numerosas fracturas y deformidades secundarias a la gran fragilidad ósea. La presentación, evidentemente, depende del tipo y subtipo de OI de cada paciente6.
Los cuadros congénitos y severos se caracterizan por fracturas in útero y durante el parto, con complicaciones serias a nivel encefálico y pulmonar, generalmente fatales. Los cuadros leves y moderados, son la mayor parte de los pacientes y corresponden al tipo I de OI, presentando pocas fracturas, deformidades y complicaciones.
La consolidación ósea no se encuentra alterada, por lo que se logra en tiempos similares a los pacientes sanos.
A consecuencia de las numerosas fracturas y de la tracción muscular se produce deformación de los huesos largos, que puede llegar a ser muy marcada en los casos más severos. Los pacientes con OI leves habitualmente no presentan mayores deformidades debido al menor número de fracturas y a la menor fragilidad ósea. Lo contrario ocurre en los pacientes con OI severa, en los que las múltiples fracturas y microfracturas producen retardo de crecimiento, encontrándose habitualmente bajo el percentil 30.
Las deformidades óseas más comunes son:
- -
Fémur: antecurvatum y varo. Es el hueso más frecuentemente fracturado y presenta una deformidad en “callado de pastor”. Además, puede presentar coxa vara y protrusión acetabular.
- -
Tibia: antecurvatum y valgo.
- -
Húmero: angulado lateralmente.
- -
Antebrazo se encuentra con mínima pronación y presenta luxación de la cabeza del radio por acortamiento del cúbito.
- -
Columna: 80-90% de los pacientes con OI severa presentan escoliosis, con severas y progresivas curvas de difícil manejo, debido a las fracturas, osteoporosis e hiperlaxitud. En casos moderados la escoliosis se presenta en el 40%.
- -
En columna cervical se puede producir impresión basilar en cerca del 2% de los pacientes (más frecuente OI tipo IV).
Otras manifestaciones de la OI en tejidos blandos
- -
Laxitud ligamentosa aumentada.
- -
Problemas cardíacos por falla de válvulas.
- -
Problemas pulmonares por mala mecánica y falla del tejido pulmonar por colágeno defectuoso.
- -
Hipoacusia: puede estar presente, pero no es una constante en los pacientes con OI. Se desarrolla con mayor frecuencia después de la adolescencia. Se produce por alteraciones a nivel de la conducción sonora en el oído medio (otoesclerosis), por alteraciones de tipo neurosensorial del oído interno o una combinación de ambas.
Presentan una fascie característica. Poseen una frente amplia con grandes huesos temporales y parietales, lo que genera una cara de tipo triangular.
Las escleras azules son indicadoras de la variante de OI dominante, si bien no es algo exclusivo y absolutamente constante de este grupo. La tonalidad azul está dada por pigmentos intraoculares que se logran observar por la delgadez de la esclera determinada por la alteración del colágeno.
Debido a las múltiples fracturas y a la inmovilización que ellas conllevan, se produce atrofia muscular que puede dificultar la rehabilitación y futura función de los pacientes. Junto a esto presentan piel delgada y fragilidad capilar.
La dentadura está comprometida en algunas variantes de la enfermedad, debido a alteraciones de la dentina. Presentan fragilidad dental, con predisposición a las fracturas y formación de caries. Son identificables por el color café o azul opalescente. El compromiso dental en la OI se presenta en los tipos I B y IV B, y puede estar presente en los tipo III.
Con respecto a la expectativa de vida, los pacientes con OI tipo I, tienen longevidad normal. Las fracturas comienzan cuando el niño comienza a caminar y presenta fragilidad ósea durante toda la vida. Hacia la adolescencia las fracturas comienzan a disminuir, pero pueden recurrir posterior a periodos de reposo, post-parto y hacia la menopausia7.
En OI tipo II, la sobrevida del Recién Nacido depende de la integridad del tórax y del cráneo. Al nacer, el niño presenta múltiples fracturas en distintas etapas de consolidación. Los huesos están severamente osteoporóticos, deformados y a menudo sin hueso cortical. Las vertebras están aplanadas.
En el resto de los tipos moderados, con tratamiento adecuado, tienen buena expectativa de vida.
5Hallazgos radiográficosEn las radiografías se observan algunas características propias de la menor densidad ósea, como también deformidades por tracción muscular y por fracturas a repetición en el lugar de mayor deformidad del hueso largo.
Lo más común es observar:
- •
Metáfisis ensanchadas
- •
Epífisis con imágenes tipo “pop corn” (tipo III)
- •
Costillas delgadas poco calcificadas
- •
Compresiones vertebrales
- •
Cráneo: hueso wormiano (60%)
- •
Huesos largos:


Lo fundamental es identificar los distintos cuadros que presentan fragilidad ósea en la infancia, lo cual puede presentar en ocasiones gran dificultad. Algunas patologías que cursan con osteoporosis son la osteoporosis idiopática juvenil, algunas neoplasias de la adolescencia (como la leucemia), tirotoxicosis, enfermedad de Cushing, etc. Junto a esto nunca se debe olvidar en pacientes con numerosas y “espontáneas” fracturas un diagnóstico muchas veces olvidado, el maltrato infantil.
7TratamientoEl tratamiento está enfocado a maximizar la función, disminuir el riesgo de fracturas, evitar la deformidad y optimizar la adaptación del niño a las actividades de la vida diaria. Para esto es necesario darle un apoyo integral y multidisciplinario.
En general, las formas leves sólo requieren manejo de las fracturas, en cambio en las más severas requieren manejo médico y quirúrgico permanente para corregir las deformidades que se van generando.
El tratamiento médico está compuesto por medicamentos, apoyo de órtesis y rehabilitación.
7.1A.-Tratamiento médicoSe han probado numerosos métodos para aumentar la masa ósea y disminuir la frecuencia de fracturas, entre ellos el fluoruro de sodio, calcitonina, esteroides anabólicos, flavonoides, vitamina C y D, hormona de crecimiento humana recombinante, sin que ninguno de ellos haya demostrado efectos significativos en la historia natural de la enfermedad.
Excepción a esto es el uso de bifosfonatos endovenosos, que han mostrado resultados alentadores en cuanto a mejorar masa ósea y reducir frecuencia de fracturas.
7.1.1i. BifosfonatosSon análogos sintéticos del pirofosfato que se unen a la hidroxiapatita en el hueso y son potentes inhibidores de la reabsorción ósea mediada por osteoclastos. Su uso en osteoporosis en adultos está bien documentado. Desde hace pocos años se ha iniciado su uso en niños.
Actualmente el más usado es el pamidronato endovenoso a intervalos de 4 a 6 meses.
Los resultados a la fecha sugieren que el pamidronato es un tratamiento efectivo para las formas severas de OI, en especial cuando se inicia precozmente. Aunque no interviene en la causa genética de la enfermedad, si mejora la historia natural y la calidad de vida de los pacientes.
De igual manera, se usa el alendronato en forma oral semanal, el que también ha demostrado mejorar la densidad ósea.
También se están usando otros bifosfonatos más nuevos como el zoledronato y risendronato8,9.
Queda por demostrar la eficacia y seguridad del tratamiento a largo plazo, la duración del tratamiento, el uso de otros bifosfonados orales, y su uso en niños y adultos con OI más leves como la tipo I.
7.1.2ii. Suplementos- a.
Calcio
- b.
Vitamina D
- c.
Hormona de crecimiento
En general, las fracturas en pacientes con OI leves se tratan igual que en el resto de la población, pero con períodos de inmovilización lo más cortos posibles, de manera de no producir fragilidad ósea por desuso.
En fracturas de huesos largos se puede utilizar un enclavijado endomedular, idealmente con sistema telescópico, para reducir la fractura y dar soporte para prevenir deformidades. Debe evitarse el uso de placas y tornillos ya que la delagada cortical no es capaz de soportarlos.
En el caso de fracturas en huesos de forma muy alterada, puede aprovecharse la oportunidad de la fractura para realizar la correción.
7.2.2B. Deformidades:El tratamiento de las deformidades de los huesos largos es de vital importancia para evitar fracturas de éstos, al alinearlos y dejarlos en condiciones mecánicas más favorables10,11.
La corrección se realiza mediante osteotomías correctoras a varios niveles (osteotomías en rosario) y se fijan mediante clavos según la técnica descrita por Sofield y Millar en 1959. Estas se fijan con clavos estáticos como agujas de Kirshner, TENS o clavos de Rush, o mediante clavos telescópicos. Estos últimos, son el tratamiento de elección y tienen la ventaja de alargarse junto con el crecimiento del niño. Se anclan a las epífisis superior e inferior12.
El clavo Dubow-Bailey fue el primero en el mercado, teniendo buenos resultados, pero se han descrito complicaciones como migración y aflojamiento de sus componentes.
Fassier desarrolló una técnica mínimamente invasiva usando el clavo Fassier-Duval. Esta se realiza a través de una incisión proximal y depende del grado de deformidad la necesidad de otra u otras en el ápice de la curva a corregir. Desde proximal se introducen los componentes macho y hembra13. Las osteotomías se realizan en forma percutánea con pequeñas incisiones en la piel (Figura 4).

La inmovilización post operatoria debe ser, lo más breve posible para prevenir osteoporosis por la inmovilización. No se utiliza yeso pelvipedio a menos que las osteotomías estén muy inestables.
El hueso que con mayor frecuencia requiere de corrección, es el fémur, seguido de la tibia. Con menor frecuencia se realiza en extremidades superiores.
Son dos las indicaciones para el enclavijado de las extremidades superiores: dificultad funcional debido a la deformidad y fracturas a repetición si limitan el uso de bastones o andador, generando problemas con la movilidad. Los antebrazos se enclavijan con agujas de Kirschner, mientras en el húmero se pueden utilizar clavos telescópicos.
Las complicaciones más frecuentes son: migración del implante, fractura del implante y no telescopaje.
Los resultados al comparar clavos telescópicos versus fijos han demostrado una tasa de entre 40 y 50% de re-operación para los fijos, y entre un 20 y 40% para los telescópicos. Con respecto a las complicaciones, se reportan tasas de 50 y 72% respectivamente11,14,15.
En cuanto a la columna, el uso de corset no es útil en OI debido a que una caja torácica frágil no permite una transferencia efectiva de la presión del corset hacia la columna vertebral y además la presión externa puede empeorar las deformidades del tórax.
Las indicaciones quirúrgicas son: escoliosis progresiva con curva mayor a 45° en formas leves de OI y mayor de 30-35° en las formas severas. Debido a que en OI tipo III y IV el crecimiento del tronco es limitado, la cirugía puede realizarse a los 7-8 años de vida.
Es importante recordar el alto riesgo anestésico de los pacientes con OI severa debido a problemas de vía aérea, enfermedad pulmonar restrictiva, impresión basilar, intubación difícil, riesgo de fracturas durante las manipulaciones y un mayor riesgo de hipertermia maligna.
7.2.3C.-RehabilitaciónEs fundamental un adecuado plan de rehabilitación en los niños con OI, en especial con el uso actual de pamidronato que ha disminuido la fragilidad ósea y ha mejorado el pronóstico para la bipedestación.
Los niños con OI pueden presentar:
- -
Deformidad de huesos largos
- -
Compresión vertebral
- -
Deformidades de columna
- -
Debilidad muscular por desuso
- -
Plagiocefalia
- -
Caderas con flexión y rotación externa
- -
Pie equino
Estos problemas pueden limitar la adquisición de destrezas motoras, en especial el control de cabeza y tronco, sentarse, gatear, levantarse y caminar. La sobreprotección de los padres también limita la independencia funcional.
Los objetivos de la rehabilitación son:
- -
Promover el desarrollo motor
- -
Facilitar las formas seguras de movimiento
- -
Maximizar la independencia funcional y así su calidad de vida
Las estrategias de rehabilitación van cambiando con la edad; en la infancia precoz los niños deben ser movilizados con especial cuidado, con cambios de posición permanentes y evitar los movimientos rotacionales debido al riesgo de fracturas. Al iniciar la marcha se les debe apoyar con buena rehabilitación para mejorar la coordinación y la fuerza muscular de modo de minimizar el riesgo de caídas. En ocasiones se pueden utilizar órtesis protectoras para prevenir deformidades y contracturas.
Como forma de mejorar la densidad ósea se sugiere realizar ejercicios de bajo impacto, actividades en agua, siempre teniendo en cuenta las restricciones deportivas que deben tener, dependiendo del tipo de OI y las condiciones de cada caso.
8Nuevos tratamientos en estudio:El trasplante de médula ósea también se encuentra en estudio al aportar stem cells que son precursores de osteoblastos. Así se produce un mosaicismo de osteoblastos normales y anormales. Los resultados de este tratamiento son controversiales debido a la dificultad para aislar los osteoblastos y el uso de inmunosupresión.
Otra estrategia en estudio, es la modificación genética de osteoblastos defectuosos extraídos del paciente para su posterior reinfusión. Esta y otras técnicas genéticas se encuentran en estudios muy preliminares y su uso en humanos aún está lejano.
Otras terapias en estado experimental16,17 pero prometedor, son:
- •
Denosumab: Anticuerpos anti RANK (receptor activator of nuclear factor-κB). Hay Algunos ensayos clinicos pequeños prometedores, pero aún en etapa experimental.
- •
Hormona de crecimento: tiene poco efecto en el crecimiento y no se ha descrito efecto en el riesgo de fracturas.
- •
Hormona paratiroidea recombinante.
- •
Anticuerpos anti sclerotina.
- •
Transplante células mesenquimales (prenatal y postnatal).
Ha habido avances muy significativos en el tratamiento y la patogenia de la OI. Se han descrito nuevos tipos de OI no colagenosas que han ampliado el campo de estudio de esta patología. El enfoque moderno es multidisciplinario e incluye tratamiento con bifosfonatos tempranamente, cirugías ortopédicas periódicas programadas y rehabilitación. Esto ha mejorado la historia natural y la calidad de vida de los pacientes. Lo que queda por desarrollar son las terapias genéticas que serán la solución definitiva a esta enfermedad.
Por último, pero de la mayor importancia, es el soporte psicológico al paciente y su grupo familiar, a través de educación y terapia de apoyo18. Existen hoy grupos de apoyo entre familas de niños con OI, donde intercambian experiencias y se ayudan entre ellas.