





Las fallas en el control de las respuestas inmunes fisiológicas o en la mantención de la tolerancia a lo propio, produce enfermedades en las cuales el mecanismo patogénico primario es inmunológico. Estas respuestas inmunes descontroladas se llaman mecanismo de hipersensibilidad y serán revisadas en este artículo, basándonos en las clasificaciones de Gell y Coombs y la más reciente de los mecanismos de daño mediados por células.
Failure to control physiologic immune responses or to maintain self-tolerance leads to diseases in wich the primary pathogenic mechanism is immunologic. These uncontrolled immune reactions are called hypersensitivity responses, and are reviewed in this article, based upon the Gell and Coombs and the more recent cell-mediated hypersensitivity reactions classifications.
Las respuestas inmunes normales, tanto celulares como humorales, nos permiten el reconocimiento de lo propio y la eliminación de patógenos. Cuando estas respuestas son exageradas, o se producen frente a sustancias normalmente inocuas, gatillan enfermedades, y las denominamos mecanismos de daño o mecanismos de hipersensibilidad (1). En el presente artículo se revisarán estos mecanismos, basados en la clasificación de Gell y Coombs, y la más reciente de Pichler (Tabla 1). No se debe olvidar que estas clasificaciones son una sistematización con fines académicos, pero que en cada patología específica, varios de estos mecanismos pueden superponerse.
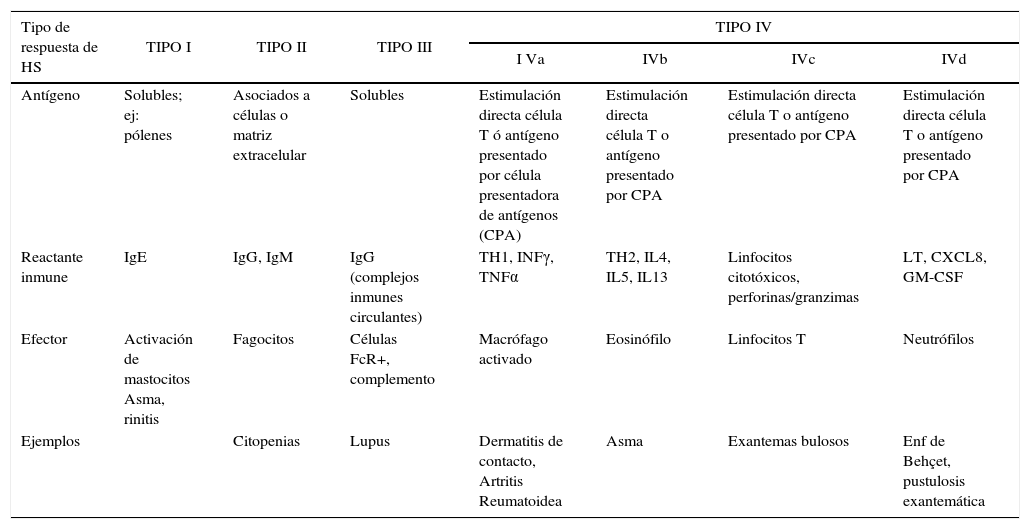
Resumen de los tipos de mecanismos de hipersensibilidad (HS)
| Tipo de respuesta de HS | TIPO I | TIPO II | TIPO III | TIPO IV | |||
|---|---|---|---|---|---|---|---|
| I Va | IVb | IVc | IVd | ||||
| Antígeno | Solubles; ej: pólenes | Asociados a células o matriz extracelular | Solubles | Estimulación directa célula T ó antígeno presentado por célula presentadora de antígenos (CPA) | Estimulación directa célula T o antígeno presentado por CPA | Estimulación directa célula T o antígeno presentado por CPA | Estimulación directa célula T o antígeno presentado por CPA |
| Reactante inmune | IgE | IgG, IgM | IgG (complejos inmunes circulantes) | TH1, INFγ, TNFα | TH2, IL4, IL5, IL13 | Linfocitos citotóxicos, perforinas/granzimas | LT, CXCL8, GM-CSF |
| Efector | Activación de mastocitos Asma, rinitis | Fagocitos | Células FcR+, complemento | Macrófago activado | Eosinófilo | Linfocitos T | Neutrófilos |
| Ejemplos | Citopenias | Lupus | Dermatitis de contacto, Artritis Reumatoidea | Asma | Exantemas bulosos | Enf de Behçet, pustulosis exantemática | |
En toda respuesta inmunológica (normal o patológica), se requiere de una fase de sensibilización, que siempre es silente. Durante esta fase, las células presentadoras de antígenos procesan los antígenos propios o extraños y los presentan a los linfocitos T CD4. Estos linfocitos serán los encargados de dirigir el tipo de respuesta inmune, ya sea de predominio celular o humoral contra este antígeno, en forma silente, hasta que al sobrepasar un determinado umbral, se desencadena el daño inmunológico y la sintomatología clínica.
Mecanismo de daño tipo IEste mecanismo es el que se observa de preferencia en las enfermedades alérgicas como rinitis o asma, donde los antígenos se denominan alérgenos: sustancias naturales, que ingresan al organismo por vías naturales y que son inocuas en una población normal. Algunos medicamentos también pueden causar este tipo de reacciones, siendo un ejemplo clásico la penicilina, cuyo metabolito peniciloil, unido a una proteína plasmática (conjugado hapteno-carrier), es capaz de inducir síntesis de IgE (1).
De acuerdo con la clasificación de Gell y Coombs, esta respuesta corresponde a un mecanismo de daño mediado por linfocitos TH2 e inmunoglobulina E (IgE), también conocida como respuesta de hipersensibilidad inmediata. Durante la fase de sensibilización, se sintetiza IgE contra alérgenos, la cual se adosará a sus receptores en la superficie de mastocitos (fase silente). Cuando el nivel de IgE en los mastocitos alcance un nivel crítico, la siguiente exposición al alérgeno originará un entrecruzamiento de los receptores de IgE (FcεRI), que llevará a su degranulación. Son las sustancias liberadas durante este proceso, las responsables de síntomas y signos como el broncoespasmo, los estornudos, la rinorrea, la congestión nasal, urticaria y angioedema, cólicos abdominales, diarrea, hipotensión y en los casos más graves, shock anafiláctico.
Esta facilidad anormal de sintetizar IgE frente a alérgenos ambientales, se denomina atopia. Esta condición viene determinada genéticamente, y se observa en aproximadamente un 20% de la población (1).
Activación de linfocitos TH2Luego del ingreso al organismo, los alérgenos son captados por células dendríticas (ubicadas en epitelio nasal, bronquial, tejido linfoide de mucosas digestivas, etc.). Estas células procesan los antígenos en su interior, migran hacia los linfonodos regionales, donde presentan los péptidos derivados del alérgeno en una molécula del complejo mayor de histocompatibilidad clase II a un linocito T naïve. Este proceso también ocurre en las mucosas respiratorias de pacientes asmáticos, y digestiva de pacientes con alergia alimentaria (2). La presencia de interleuquina 4 (IL4), y la ausencia de estímulos inflamatorios de la inmunidad innata (como ocurre en los procesos infecciosos), permiten la activación de los factores de transcripción STAT6 y GATA-3. Este último es el principal regulador de la diferenciación de este linfocito hacia un fenotipo TH2, y potencia la expresión de los genes de las interleuquinas 4, 5 y 13 (IL4, IL5 e IL13). Estas citoquinas son las responsables de que las células plasmáticas (linfocitos B) que reconocen el mismo alérgeno, hagan un cambio en el isotipo de cadenas pesadas de las inmunoglobulinas que secretan y comiencen a producir IgE. Además, la IL5 juega un rol importante en la activación y quimiotaxis de eosinófilos, y la IL13 es capaz de estimular la hipersecreción mucosa bronquial.
IgE/ ReceptoresLa IgE así sintetizada es específica para el alérgeno gatillante. Un individuo puede producir IgE específicas para uno o varios alérgenos simultáneamente. Esta IgE sale a circulación y rápidamente se une a receptores específicos de alta afnidad, los FcεRI, ubicados en la superficie de mastocitos titulares y basófilos. Una vez unida a su receptor, la IgE está preparada para cumplir con su función de receptor específico para el alérgeno, y las células quedan así sensibilizadas y preparadas para reaccionar frente a un próximo encuentro con el antígeno. Cuando esto ocurre, el entrecruzamiento de receptores FcεRI que se unen a un alérgeno polivalente, permite que sus porciones intracelulares, acopladas a tirosin kinasas activen una cascada de señales hacia el intracelular, que culminan en la degranulación de las células efectoras (1, 2).
Fases de la respuestaLas células efectoras del daño en la hipersensibilidad inmediata son los mastocitos, basófilos y eosinófilos, que comparten la característica común de poseer gránulos citoplasmáticos ricos en mediadores preformados y su capacidad sintetizar de novo mediadores lipídicos y citoquinas.
La primera fase de las respuestas de hipersensibilidad mediadas por IgE (denominada reacción inmediata), tiene características clínicas bien definidas: eritema, edema y prurito cutáneos; estornudos y rinorrea; tos, broncoespasmo, edema y secreción mucosa en el tracto respiratorio inferior; náuseas, vómitos, diarrea y cólicos, e hipotensión. Todos estos síntomas son inducidos por los mediadores preformados (histamina, triptasa, proteoglicanos) y los mediadores lipídicos (prostaglandinas y leucotrienos). Pasadas 6 a 24 horas puede producirse una segunda ola de síntomas y signos, conocida como la reacción de fase tardía de las respuestas de hipersensibilidad tipo I. Esta se caracteriza por el edema e influjo de leucocitos, por acción de citoquinas sintetizadas de novo (IL1, IL3, IL4, IL5, IL6, IL13 y factores de crecimiento) y liberadas varias horas después de que los mastocitos y basófilos se han activado. Su rol fundamental es la amplificación de la respuesta inflamatoria alérgica (1-3).
Cuando el estímulo antigénico es persistente (como puede ser la exposición continua a alérgenos ambientales como los ácaros del polvo de habitación), esta respuesta se transforma en una respuesta infamatoria crónica, donde predomina la infltración eosinofílica, el daño a los tejidos y su reparación por tejido fbrótico (remodelación de la vía aérea en el asma). Siguiendo la nueva clasifcación por mecanismos inmunes involucrados, esta fase de las respuestas TH2 corresponde a un mecanismo de hipersensibilidad tipo IVb (ver más adelante).
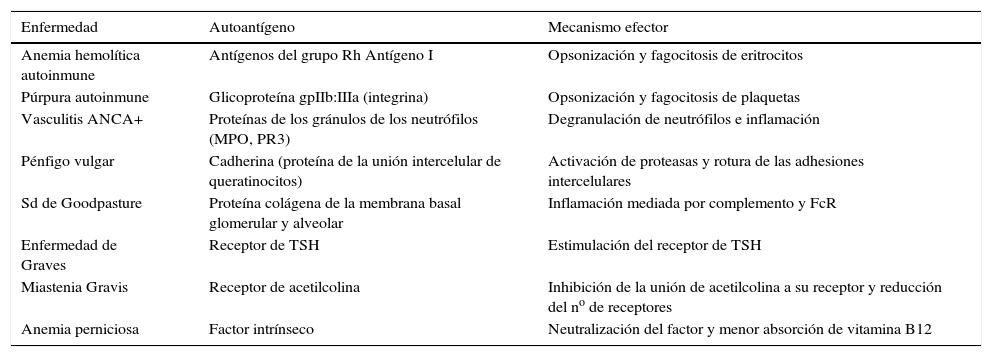
Mecanismo de daño tipo IIEste mecanismo de daño es el que se observa preferentemente en las citopenias autoinmunes y en algunas enfermedades autoinmunes órga-no-específcas (Tabla 2). Corresponde a un mecanismo de daño de tipo humoral, mediado por inmunoglobulinas de tipo G ó M (IgG e IgM), con capacidad de opsonizar, reclutar leucocitos y de activar el complemento, gatillando así respuestas inflamatorias, o de inducir cambios funcionales a nivel de receptores celulares (4).
Ejemplos y mecanismos de daño en enfermedades mediadas por un mecanismo de hs tipo II
| Enfermedad | Autoantígeno | Mecanismo efector |
|---|---|---|
| Anemia hemolítica autoinmune | Antígenos del grupo Rh Antígeno I | Opsonización y fagocitosis de eritrocitos |
| Púrpura autoinmune | Glicoproteína gpIIb:IIIa (integrina) | Opsonización y fagocitosis de plaquetas |
| Vasculitis ANCA+ | Proteínas de los gránulos de los neutrófilos (MPO, PR3) | Degranulación de neutrófilos e inflamación |
| Pénfigo vulgar | Cadherina (proteína de la unión intercelular de queratinocitos) | Activación de proteasas y rotura de las adhesiones intercelulares |
| Sd de Goodpasture | Proteína colágena de la membrana basal glomerular y alveolar | Inflamación mediada por complemento y FcR |
| Enfermedad de Graves | Receptor de TSH | Estimulación del receptor de TSH |
| Miastenia Gravis | Receptor de acetilcolina | Inhibición de la unión de acetilcolina a su receptor y reducción del no de receptores |
| Anemia perniciosa | Factor intrínseco | Neutralización del factor y menor absorción de vitamina B12 |
(adaptado de 4)
Como ejemplo de enfermedades donde podemos observar este mecanismo de daño, sin un autoantígeno involucrado podemos citar la fiebre reumática y algunas citopenias por fármacos. En el caso de la fiebre reumática, se produce una reacción cruzada de los anticuerpos que reaccionan contra la pared celular del estreptococo y reconocen un antígeno miocárdico por mimetismo molecular. En el caso de los fármacos, las drogas o metabolitos de las mismas, unidos a proteínas de la membrana celular, actúan como haptenos, e inducen la síntesis de anticuerpos (4, 5).
Mecanismos efectoresLas inmunoglobulinas se adosarán a las superficies celulares donde se encuentran los antígenos (opsonización). Este es el punto de partida que desencadena el daño.
Citopenias (4)- -
Los fragmentos Fc de las IgG serán reconocidos por receptores específicos de alta afnidad (FcγRI) ubicados principalmente en células macrofágicas. De este modo, los glóbulos rojos, neutrófilos o plaquetas serán sacados de circulación cuando pasen a través del sistema retículo-endotelial en el hígado y el bazo. Clínicamente se traducirá en anemia hemolítica, neutropenia o plaquetopenia, dependiendo de la célula blanco.
- -
Las inmunoglobulinas también pueden activar el complemento, cuyos productos de activación también opsonizan las células, favoreciendo su remoción de la circulación. Menos frecuente es la lisis intravascular por activación del complemento.
- -
Las inmunoglobulinas unidas a sus células blanco, activarán localmente al complemento por la vía clásica, produciéndose por una parte fragmentos opsonizantes como C3b y C4b, y por otra, fragmentos pro-inflamatorios como C3a y C5a (anafilotoxinas). Estas últimas regulan la vasodilatación e incrementan la permeabilidad capilar tanto en forma directa, como estimulando la liberación de histamina en basófilos y mastocitos. Además estimulan el estallido respiratorio en macrófagos, neutrófilos y eosinófilos. El C3a modula la síntesis de citoquinas pro-inflamatorias (IL6 y factor de necrosis tumoral alfa (TNFα)), y el C5a es un poderoso quimioatractante de macrófagos y polimorfonucleares (4,6).
- -
Tanto los fragmentos Fc de las inmunoglobulinas, como los productos de la activación del complemento son reconocidos por receptores específicos en la superficie de macrófagos y neutrófilos, lo que llevará a su activación y degranulación (al tratarse de antígenos titulares, el tamaño impide la fagocitosis de las células, por lo que los leucocitos liberan todo el contenido de sus gránulos sobre los tejidos).
- -
La primera consecuencia de esto es el daño tisular por la acción de las proteasas y las especies reactivas del oxígeno liberadas al medio. En este proceso también se producen una serie de factores quimiotácticos, los cuales atraerán a un mayor número de células inflamatorias al tejido blanco, amplificando así el daño (7).
- -
Como se aprecia en la Tabla 2, cuando los anticuerpos ejercen acciones funcionales sobre determinados receptores (bloqueo del receptor de acetilcolina en la miastenia gravis o estimulación de receptores de TSH en la enfermedad de Graves), no hay reacciones inflamatorias ni daño tisular.
En este tipo de respuesta de hipersensibilidad, las inmunoglobulinas forman complejos inmunes junto con el antígeno (propio o extraño), y a diferencia del mecanismo anterior, estos complejos se encuentran en circulación. El daño tisular dependerá entonces de los sitios donde estos inmunocomplejos se depositen, y no del origen del antígeno gatillante. La enfermedad más emblemática de este tipo de daño, es el lupus eritematoso sistémico: se generan autoanticuerpos que reconocen proteínas tan ubicuas como el ADN o nucleoproteínas, y las manifestaciones clínicas principales son las artritis, serositis, nefritis y vasculitis. Como ejemplo de antígenos exógenos, destacan las vasculitis asociadas a la infección por virus de la hepatitis B, donde los inmunocomplejos están formados por antígeno de superficie del virus y anticuerpos anti antígeno de superficie. Algunos fármacos también son capaces de generar reacciones adversas por este mecanismo, manifestándose como una enfermedad del suero o una vasculitis por hipersensibilidad: cefaclor, cefalexina, trimetropim-sulfametoxazol, amoxicilina, anti-inflamatorios no esteroidales, diuréticos y algunos biológicos (4, 5).
Factores que favorecen el depósito de complejos inmunesSi bien en toda respuesta inmune normal se generan inmunocomplejos, éstos son rápidamente retirados de la circulación. El daño se genera cuando estos complejos inmunes se forman en cantidades excesivas, que sobrepasan la capacidad normal de eliminación. Otros elementos importantes en la patogenicidad de estos complejos inmunes son el tamaño (los complejos más grandes son rápidamente fagocitados, mientras que los de menor tamaño tienen mayor tendencia a depositarse), y la presencia de antígenos catiónicos dentro del complejo (éstos se unen con mayor avidez a los componentes aniónicos de las membranas basales glomerulares y de los vasos sanguíneos). Las características del fujo sanguíneo también son importantes en la patogenicidad: estos complejos inmunes se depositan de preferencia en los sitios donde se produce un ultrafiltrado (glomérulos y sinoviales).
La eliminación normal de complejos inmunes circulantes ocurre en el sistema retículo-endotelial, donde las células macrofágicas los captan, reconociendo las porciones Fc de las inmunoglobulinas, o los fragmentos opsonizantes del complemento a través de receptores específicos. Mutaciones tanto en el complemento como en los receptores de inmunoglobulinas, pueden llevar a disminuir su clearence, favoreciendo el posterior depósito (8).
Mecanismos efectoresUna vez depositados en el endotelio, los complejos inmunes activarán las cascadas de la coagulación y del complemento, activando así a los leucocitos de un modo similar a lo descrito en el mecanismo de tipo II: producción de anfilotoxinas, principalmente de C5a, que llevará a una activación endotelial (ya que estimula a los mastocitos a liberar aminas vasoactivas) y aumento de la permeabilidad capilar, favoreciendo un mayor depósito de complejos y aumentando el flujo de polimorfonucleares a la zona. Estas células activadas liberarán enzimas, mediadores lipídicos y citoquinas que terminarán por dañar los vasos sanguíneos, manifestándose clínicamente como una vasculitis, artritis, nefritis o serositis (Figura 1).
.")
Depósito de complejos inmunes en la pared del endotelio, que activan la cascada de la coagulación y el complemento, generando sustancias quimiotácticas para neutrófilos. Estos dañan directamente las paredes vasculares por acción de sus enzimas. (adaptado de 12).
Este tipo de respuesta de hipersensibilidad, también conocida como respuesta de hipersensibilidad retardada, involucra mecanismos celulares de daño. Tradicionalmente descrita como reacciones linfocitarias que llevan a la activación de macrófagos y formación de granulomas, como en el caso de la tuberculosis, o a acciones citotóxicas directas de linfocitos T CD8 (4), como en el caso las hepatitis virales, su descripción fue ampliada por Pichler, quien la clasificó en cuatro subtipos (Tabla 1), de acuerdo con las observaciones clínico-patológicas de las reacciones adversas a fármacos (5).
En todos los casos, el antígeno es inicialmente captado por células presentadoras de antígenos, quienes lo procesan y presentan en el contexto de moléculas de histocompatibilidad a linfocitos T en los linfonodos regionales, durante la fase silente de sensibilización. Son los diferentes tipos de linfocitos T, mediante patrones específicos de citoquinas, quienes dirigen el tipo de inflamación que se produce.
Reacciones tipo IVaEstas reacciones involucran la activación de linfocitos TH1, los cuales producen grandes cantidades de interferón gamma (INFγ), principal citoquina activadora de macrófagos. Estas últimas células liberan enzimas lisozomales, especies reactivas del oxígeno, óxido nítrico y más citoquinas pro-inflamatorias (como TNFα, IL1), lo cual daña el tejido localmente y atrae un rico infiltrado celular de neutrófilos y monocitos. Frecuentemente estas reacciones son crónicas, y los tejidos dañados son reemplazados por tejido conectivo, llevando a la fibrosis. El INFγ también estimula a linfocitos CD8 en sus actividades citotóxicas, por lo que lo habitual es encontrar una combinación de reacciones tipo IVa con IVc, como en el caso de las dermatitis de contacto (4, 5). Si bien en artritis reumatoide son múltiples los tipos de células y citoquinas involucradas en el daño articular, la base de la destrucción es la infamación mediada por macrófagos (9), activados por las células T (Figura 2).
Reacciones tipo IVb
Estas corresponden a la fase tardía de las respuestas inmunes mediadas por linfocitos TH2 (ver mecanismo de daño tipo I): la elevada tasa de IL5 producida induce una inflamación eosinofílica en los tejidos. Este tipo de reacciones alérgicas es el que se observa en asma y rinitis alérgicas, en dermatitis atópica y en algunos tipos de exantemas maculopapulares inducidos por fármacos. En el síndrome de Churg Strauss, este mecanismo tiene un rol muy importante en el daño y en la hipereosinofilia característica del cuadro, sin embargo, al igual que en otras mesenquimopatías, no constituye un mecanismo único para explicar la patogenia: en este cuadro también encontramos células T con perfil TH1, TH17, y daño mediado por ANCA (11).
EosinófilosEstos granulocitos maduran en la médula ósea con el estímulo de citoquinas como IL5, IL3 y GM-SCF, y su localización habitual es en las mucosas respiratoria, digestiva y urinaria. Es en estas mismas zonas donde pueden activarse y aumentar su número, en el contexto de reacciones inflamatorias alérgicas (fase tardía de las reacciones de hipersensibilidad inmediata), atraídos sobretodo por la eotaxina y el aumento de expresión de moléculas de adhesión, inducidos por IL4 e IL13 (1, 2). Tienen dos tipos de gránulos: específicos y primarios. Los gránulos específicos son ricos en proteínas catiónicas como la proteína básica mayor (PBM), peroxidasa eosinofílica, proteína catiónica eosinofílica y la neurotoxina eosinofílica. Los gránulos primarios son similares a los de otros granulocitos, y contiene los cristales de Charcot-Leyden. No existe consenso respecto de los estímulos que gatillarían su degranulación. Además, tienen cuerpos lipídicos intracitoplasmáticos (carentes de membrana), principal sitio de síntesis de eicosanoides (2). La proteína catiónica no solo posee una actividad citotóxica directa: también estimula la producción de factor de crecimiento transformante beta, la principal citoquina pro-fibrótica. Las otras proteínas catiónicas también son capaces de estimular la síntesis de moléculas profibróticas. De este modo, el eosinófilo y sus mediadores juegan un rol en la injuria epitelial, engrosamiento de las membranas basales e hipertrofia de musculatura lisa (10).
Reacciones tipo IVcEn este tipo de reacciones, son los propios linfocitos (CD4 y CD8) los efectores del daño: por sus acciones citotóxicas, mediadas por perforinas y granzimas y por contacto de moléculas Fas y FasL, lisan diversos tipos celulares como queratinocitos o hepatocitos. Este mecanismo es clave en varios tipos de enfermedades ampollares, como la necrolisis epidérmica tóxica por fármacos (4, 5).
Reacciones tipo IVdCorresponde a inflamaciones neutrofílicas, como las observadas en las pustulosis exantemáticas agudas (inducida por fármacos), o la enfermedad de Behçet. Los linfocitos producen la quimioquina CXCL8 que atrae neutrófilos y GM-CSF que evita su apoptosis (4, 5).
ConclusionesEn las enfermedades con una patogenia primariamente inmunológica, los mecanismos que llevan al daño celular y tisular, pueden ser predominantemente de tipo celular o de tipo humoral. En estos dos grupos principales de respuestas inmunes, las características del tipo de célula involucrada o del isotipo de inmunoglobulina que ejercerá el daño dependerán del patrón de citoquinas producidas por los linfocitos T sensibilizados al antígeno gatillante.
La autora declara no tener confictos de interés, con relación a este artículo.