



Las enfermedades autoinmunes son patologías de gran complejidad clínica, difícil diagnóstico y complejo tratamiento cuya etiología permanece aún desconocida pese a los múltiples avances realizados en los últimos años. En la génesis de estas enfermedades participan múltiples factores que conflyen entre sí para dar origen a cada una de las patologías autoinmunes conocidas, sean estas órgano-específicas o sistémicas. Entre estos elementos se incluyen la pérdida de los mecanismos de tolerancia, factores de susceptibilidad genética (polimorfismos HLA, genes no HLA y mecanismos epigenéticos), factores ambientales (agentes vivos de enfermedad, agentes inorgánicos, hormonas y otros) y factores inmunológicos (linfocitos reguladores, citoquinas y moléculas coestimulatorias, entre otros).
La identificación de estos factores permitirá mejorar el conocimiento de los variados mecanismos que median estas complejas enfermedades, facilitando no sólo el entendimiento de su etiología sino también perfeccionar las herramientas terapéuticas para enfrentarlas.
Autoimmune diseases are pathologies of great clinical complexity, difficult diagnosis and treatment complex which etiology still remains unknowns despite the many advances made in recent years. In the genesis of these diseases involves multiple factors that converge together to give rise to each of the autoimmune diseases knows, whether organ specific or systemic. These elements include loss of tolerance mechanisms, genetic susceptibility factors (HLA polymorphisms, genes non-HLA and epigenetic mechanisms), environmental factors (living agents of disease, inorganic agents, hormones, etc.) and immunologic factors (regulators lymphocyte, cytokines, costimulatory molecules and others).
Identifying these factors will improve the knowledge of the various mechanisms that mediate these complex diseases facilitating not only the understanding of the etiology but also improve the therapeutic tools to address them.
La palabra inmunidad deriva del latín “immunitas” que era el nombre dado a los senadores romanos para expresar su condición de “protegidos” o “intocables” (1). Es por esto que cuando hablamos del sistema inmune (SI) la primera función que recordamos es la de protección contra los agentes infecciosos, pero esta no es la única ni la más importante de sus funciones cuyo verdadero y más importante objetivo es la inmunovigilancia (2).
Esta inmunovigilancia implica no sólo la protección contra los agentes externos de daño y enfermedad, sean orgánicos (bacterias, virus, hongos y parásitos) o inorgánicos (mercurio, hidrocarburos, luz ultravioleta, radiación y otros) sino también la protección contra agentes propios cuando estos son alterados por el envejecimiento o trasformadas por procesos neoplásicos (1, 2). Esto implica la existencia de un complejo mecanismo de presentación y reconocimiento antigénico, así como también de un riguroso sistema de control y regulación que permita discriminar y aceptar a lo “propio y sano” y al mismo tiempo reconocer y atacar a lo “extraño” condición que se conoce como tolerancia inmunológica (2).
Cuando la tolerancia falla nos encontramos frente a la aparición de enfermedades inmunológicas que se pueden clasificar en dos grandes grupos: Las reacciones por hipersensibilidad a agentes externos, en donde el SI reconoce como potencialmente dañino a un elemento no necesariamente peligroso para el organismo y monta una respuesta inflamatoria contra él, situación que es la base de la patogenia de las enfermedades alérgicas; así como también reacciones de hipersensibilidad contra el propio organismo situación que da origen a las llamadas enfermedades por autoinmunidad (2). Estas últimas pueden ser órgano específicas o sistémicas dependiendo del tipo y ubicación del blanco antigénico involucrado.
El término tolerancia implica la falta de respuesta a un antígeno, provocada por la exposición previa a éste (2). De este modo todo elemento que es reconocido por el SI (antígeno) puede generar dos tipos de respuesta: ser reconocido y aceptado como propio: antígeno tolerógeno, o ser reconocido pero capaz de generar una respuesta inmune defensiva: antígeno inmunógeno. A su vez la tolerancia a antígenos propios se conoce como autotolerancia.
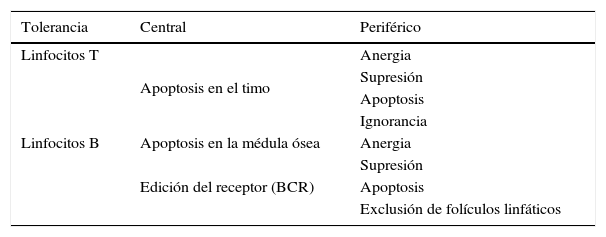
En general se puede hablar de dos tipos de tolerancia: la central que es llevada a cabo en los órganos linfoides primarios del SI (médula ósea y timo) y la periférica que es llevada a cabo en los órganos linfoides secundarios (bazo y ganglios) (2). El principal mecanismo por el que ejerce su acción la tolerancia central implica la muerte celular programada o apoptosis de aquellos linfocitos inmaduros y autoreactivos, definidos como tal porque su receptor antigénico reconoce con alta afinidad a un antígeno propio motivo por el cual son destruidos antes de madurar y salir a circulación (selección negativa) (2). Pese a lo anterior, existen linfocitos autoreactivos capaces de madurar y salir a circulación porque la afinidad con que realizan el reconocimiento de un autoantígeno es menor, permitiéndoles escapar del proceso de apoptosis; y es sobre esta población que actúan los mecanismos de tolerancia periférica entre los que se incluyen la anergia clonal, que es una inactivación funcional de linfocitos autoreactivos sin muerte celular (mecanismo que sería generado por una activación incompleta dada por la ausencia de la segunda señal o señal coestimulatoria). La supresión, que sería llevada a cabo por los llamados Linfocitos Reguladores (LR) mediante la secreción de citoquinas o vía contacto directo célula a célula; y la ignorancia inmunológica, que consiste en una ausencia de respuesta inmune cuyo mecanismo exacto aún es desconocido, pero se cree tendría relación con la presencia de antígenos crípticos u ocultos para el sistema inmune como lo conforman los antígenos oculares y testiculares (2). Ver tabla nº 1.
Mecanismos de tolerancia central y periférica para linfocitos T Y B
| Tolerancia | Central | Periférico |
|---|---|---|
| Linfocitos T | Apoptosis en el timo | Anergia |
| Supresión | ||
| Apoptosis | ||
| Ignorancia | ||
| Linfocitos B | Apoptosis en la médula ósea | Anergia |
| Supresión | ||
| Edición del receptor (BCR) | Apoptosis | |
| Exclusión de folículos linfáticos |
(adaptado de ref 2)
También se ha descrito la presencia de apoptosis en la tolerancia periférica, pero esto se considera más bien un mecanismo de finalización de respuesta y retorno a la homeostasis inicial, ya que es un mecanismo que se activa una vez que el antígeno causal de la respuesta inmune ha desaparecido (1, 2).
Considerando esta compleja regulación, se hace un poco más sencillo entender por qué la pérdida de la autotolerancia se relaciona con el desarrollo de enfermedades autoinmunes (EAI). No obstante, este hecho por sí sólo no es suficiente para el desarrollo y perpetuación de la enfermedad, ya que al igual que en otras enfermedades, su origen no solo depende del individuo sino también de las características del medio ambiente en el que el individuo está inmerso. En este punto cobra relevancia el adagio que cita: “las enfermedades complejas se deben a la conjugación de una terreno fértil, un individuo susceptible y la mala suerte”(3). Sobre los principales actores involucrados en la patogenia de las enfermedades autoinmunes es el tema a desarrollar a continuación.
1Participación de la genética en la autoinmunidadCada individuo posee una base o background genético que le confiere susceptibilidad o protección ante ciertas enfermedades, pero esta condición no es suficiente por si sola, para el inicio y desarrollo de la enfermedad (4). Estudios en gemelos homocigotos han permitido establecer que si bien existe un componente heredable en el desarrollo de estas enfermedades, éste no es el único involucrado. Las tasas de concordancia distan mucho de estos resultados y sólo en casos aislados, como la Diabetes Tipo I (DM-1) son cercanas al 50% (1, 3).
Susceptibilidad genética y Complejo Mayor de Histocompatibilidad (MHC)Los genes más frecuentemente involucrados en las EAI son aquellos que tienen relación con el MHC, que en el caso de los humanos se engloba bajo el concepto de antígenos leucocitarios humanos o HLA y que alberga a genes involucrados en los procesos de procesamiento, presentación y reconocimiento antigénico. Además destacan genes del sistema complemento (C2, C4 y factor B) y de Interleuquinas (IL) como el Factor de Necrosis Tumoral alta (TNF-a) y la Linfotoxina (3 (LT(3) (2), que a su vez también han sido señalados como partícipes en el desarrollo de autoinmunidad.
a.Genes HLALa hipótesis general consiste en que ciertos polimorfismos de estos genes codifican para proteínas que influyen negativamente en la mantención de la tolerancia o bien facilitan el desarrollo de autoinmunidad (4, 5). Conocido es el ejemplo del HLA-B27 y su asociación con el desarrollo de pelviespondilopatías (PEP), principalmente con la Espondiloartritis Anquilosante (EAA), existiendo varias teorías con respecto a esta molécula para explicar el mayor riesgo relativo a desarrollar la enfermedad entre los portadores.
Entre ellas podemos encontrar la del “péptido artritogénico”, la cual plantea que existiría similitud o mimetismo molecular entre un péptido bacteriano o viral con un péptido constitutivo del organismo de modo que la unión estereoquímica a los bolsillos de anclaje del HLA-B27 sería interpretada por los Linfocitos T Citotóxicos (LTC) como una primera señal e iniciaría el camino hacia la autoinmunidad (6, 7).
También está la teoría del “misfolging o plegamiento incompleto” de esta molécula que generaría la acumulación de la cadena pesada mal plegada en el retículo endoplásmico y la formación de homodímeros o heterodímeros de las cadenas del HLA-B27, generando mecanismos de estrés que llevan a la activación del Factor Nuclear ϰB (NFϰB) gatillando pérdida de tolerancia y autoinmunidad (6, 7).
Otra molécula señalada para conferir mayor susceptibilidad al desarrollo de enfermedades autoinmunes, es este caso para Artritis Reumatoide (AR), lo conforman los alelos del HLA DR4 que comparten una secuencia aminoacídica conocida como “epítope compartido” (QKRAA, QRRAA o RRRAA), ubicado en la tercera región hipervariable de la cadena beta de las moléculas DRB1, DR6 y DR10 (7-10). Estas moléculas se caracterizan porque uno de sus bolsillos de unión peptídica (P4) posee carga positiva, de modo que los péptidos que poseen arginina se unen ineficientemente a él ya que sus cargas se repelen, mientras que los péptidos citrulinados que poseen carga neutra, pero mantienen sus características polares, pueden unirse con mayor afinidad y ser reconocidos por los LT bajo condiciones proinflamatorias, facilitando su reconocimiento como extraños y gatillando la activación de LB autoreactivos, con generación de autoanticuerpos y daño tisular (7-9).
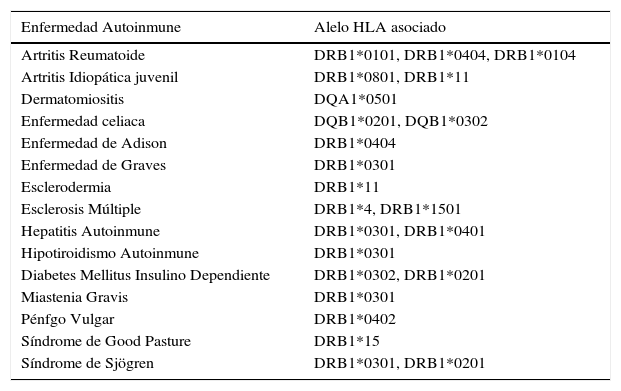
Existen evidencias de otras asociaciones entre alelos del HLA y enfermedades autoinmunes (Lupus, Kawasaki, Behcet y otras), cuyo mecanismo molecular aún no está definido, pero se cree similar a los antes descritos y cuya participación aportara nuevas respuestas en el estudio de la autoinmunidad (4, 11, 12) (Ver tabla nº 2).
Ejemplos de enfermedades autoinmunes y los alelos hla a los que han sido relacionadas
| Enfermedad Autoinmune | Alelo HLA asociado |
|---|---|
| Artritis Reumatoide | DRB1*0101, DRB1*0404, DRB1*0104 |
| Artritis Idiopática juvenil | DRB1*0801, DRB1*11 |
| Dermatomiositis | DQA1*0501 |
| Enfermedad celiaca | DQB1*0201, DQB1*0302 |
| Enfermedad de Adison | DRB1*0404 |
| Enfermedad de Graves | DRB1*0301 |
| Esclerodermia | DRB1*11 |
| Esclerosis Múltiple | DRB1*4, DRB1*1501 |
| Hepatitis Autoinmune | DRB1*0301, DRB1*0401 |
| Hipotiroidismo Autoinmune | DRB1*0301 |
| Diabetes Mellitus Insulino Dependiente | DRB1*0302, DRB1*0201 |
| Miastenia Gravis | DRB1*0301 |
| Pénfgo Vulgar | DRB1*0402 |
| Síndrome de Good Pasture | DRB1*15 |
| Síndrome de Sjögren | DRB1*0301, DRB1*0201 |
(adaptado de 41)
Así como para los genes del HLA también existen asociaciones y estudios de susceptibilidad descritos para otros genes del MHC no HLA, como los que codifican para las chaperonas Tapasinas 1 y 2, los componentes del proteosoma, la proteína de shock térmico 70 (HSP-70) (13) e Interleuquinas (IL) (14) y para genes reguladores no relacionados con el MHC como el de la proteína asociada a los linfocitos T citotóxicos 4 (CTLA-4) o CD 152 y la proteína tirosina fosfatasa, no receptor tipo 22 (linfoide) (PTPN22) (15, 16).
Se han descrito deficiencias congénitas de componentes del complemento como C1q, C2 y C4 cuyos portadores tiene un riesgo elevado de desarrollar lupus eritematoso sistémico (LES) de hasta 90% para C1q, 75% para C4 y 10% para C2 (1, 16), lo cual tendría relación con una merma en el clearance de complejos inmunes (CI) y en la eliminación de detritus celulares y células apoptóticas por falta de opsonización.
También se ha identificado polimorfismos en los genes de ILs (IL-2, 12, 18, TNF α INF-γ) con una hiperproducción de la citoquina involucrada y el consecuente desbalance hacia un ambiente proinflamatorios (13, 14), implicados no sólo en el desarrollo de autoinmunidad, sino también en la aparición de neoplasias (11).
Finalmente, en el caso de los genes reguladores, destacan los ejemplos del CTLA-4 y PTPN 22 ya que ambos codifican para proteínas que actúan como señales inhibitorias hacia los LT activados y cuyas mutaciones se traducen en la ausencia de esta señal inhibitoria facilitando la perpetuación de la respuesta inmune, el quiebre de la tolerancia y el inicio de la autoinmunidad (16).
EpigenéticaLa epigenética (del griego επ¿ que significa epi o sobre y γενετικ¿ς, que significa genética) estudia los mecanismos o factores no genéticos que intervienen en la regulación heredable de la expresión génica pero sin que se generen cambios en la secuencia de nucleótidos (3). Funcionalmente, la epigenética induce la expresión o represión de genes y es capaz de conferir una cierta plasticidad metabólica a la célula que le permite ser resiliente o “capaz de adaptarse” a los cambios ambientales (3). Los mecanismos más importantes son:
a. Metilación del ADNLa metilación es el proceso mediante al cual se agrega un grupo metilo al quinto carbono de los residuos de citosina del ADN, generando una conformación cerrada de la cromatina, lo que se traduce en el silenciamiento de genes ya que con mayor frecuencia ésta se produce en las islas CpG (regiones con alta concentración de citosina y guanina que forman parte de la región de genes promotores) inhibiendo la trascripción rio debajo de esa secuencia génica (1, 3, 16). Ejemplos fisiológicos de este mecanismo lo conforman la impronta génica y la inactivación del cromosoma X en la mujer.
Estudios recientes han demostrado que la metilación es un mecanismo de defensa contra virus y parásitos para evitar que éstos logren dañar el ADN, pero agentes ambientales también son capaces de gatillar metilación (3). Al respecto se ha visto que elementos dietarios como las piridoxinas, metionina, colina y el ácido fólico pueden estimular la metilación del ADN al ser cofactores de la ADN metiltransferasa y de este modo influir sobre la expresión de genes reguladores dando inicio a la pérdida de tolerancia y autoinmunidad (3, 16).
b. Modificación de histonasLas histonas son proteínas altamente conservadas que residen en el núcleo y pueden clasificarse en las que forman parte del core (H2A, H2B, H3 y H4) en torno a las cuales se enrolla la cromatina para dar origen al nucleosoma; y las histonas de unión (H1 y H5) (3, 16). Los nucleosomas se organizan en octámeros y de este modo se enrolla el material genético lo que implica que no es de fácil acceso a la maquinaria transcripcional.
Mediante modificaciones post traduccionales de las histonas por acetilación, fosforilación, metilación, deaminación, isomerización de prolinas y ubiquitinización puede cambiar la configuración de las histonas generando variaciones (3). La acetilación (que se relaciona con la promoción de la expresión génica) y deacetilacion (que estimularía la represión génica) en los residuos de lisina de la cola de las histonas del nucleosoma, son considerados uno de los más importantes mecanismos de regulación en la expresión de genes, de forma que combinaciones específicas de estos servirían como un código de regulación que determina si el gen debe expresarse o silenciarse (3).
Ambos mecanismos han sido indicados como potenciales causales del quiebre de la tolerancia, tanto mediante alteraciones en la metilación del ADN, como en la represión de la transcripción. Son ejemplos fisiológicos de estos mecanismos la hipometilación que posee el gen promotor del Interferón gamma (IFN-γ) en las células Th1, lo cual estimularía su transcripción, así como también una modificación de histonas de tipo represiva sobre el locus de las IL 4 y 13, situación que ocurre de forma inversa en las células Th2 (3).
En el caso de las enfermedades autoinmunes, se ha identificado hipometilación en el ADN de linfocitos T y B (LT y LB) de los pacientes con LES con la consecuente sobre expresión de algunos genes (3, 17). Este mecanismo podría explicar, por ejemplo, la aparición de lupus por drogas ya que fármacos como la Procainamida e Hidralazina inhiben la metilación del ADN (3).
2Participación del medio ambiente en la autoinmunidadAgentes vivosLa participación de agentes patógenos de enfermedad explicaría las variaciones geográficas y estacionales de algunas EAI, como lo que sucede con la enfermedad de Kawasaki (18). Los virus y las bacterias son los microorganismos que con mayor frecuencia han sido involucrados en el desarrollo de autoinmunidad, ya sea como factor causal al gatillar el proceso inflamatorio inicial o bien como factores perpetuadores del proceso inflamatorio (19, 20). Esta acción sería mediada a través de los mecanismos que detallaremos a continuación:
a. Mimetismo Molecular (MM)Consiste en la existencia de una secuencia de amino ácidos que es idéntica o muy similar entre un antígeno propio y una parte del microorganismo generando una reactividad cruzada de forma que en presencia de una segunda señal coestimulatoria (21), por ejemplo en el contexto de un ambiente proinflamatorio, LT autorreactivos podrían activarse e iniciar una respuesta inmune aberrante. Esta condición habitualmente, va de la mano de una activación policlonal de LB con generación de autoanticuerpos del tipo antinuclear o antifosfolípido entre los más frecuentes, situación que puede verse en procesos infecciosos, así como también en patología neoplásica y autoinmune (20, 21).
El ejemplo más conocido de este mecanismo lo constituye el desarrollo de la enfermedad reumática, que acontece tras la infección por un Estreptococo betahemolítico grupo A en donde el antígeno M y el antígeno carbohidrato del grupo A presentan mimetismo molecular con la miosina presente en el tejido cardíaco, así como también con otros autoantígenos del tejido sinovial, piel y sistema nervioso central, originando las manifestaciones clínicas clásicas de la enfermedad reumática (19, 21, 22), lo cual explica además la detección de autoanticuerpos contra estos antígenos en el suero de los pacientes afectados (21).
Existen en la actualidad múltiples ejemplos de MM aplicados a varios modelos de autoinmunidad: Se ha establecido una similitud en la proteínas de Citomegalovirus (CMV) y del Virus Epstein Barr (EBV) con el epitelio de las glándulas salivales, asignándoles un rol etiológico en el desarrollo del Síndrome de Sjögren (SS) (23), también con Clamidia pneumoniae y Haemophilus influenzae B (Hib) en la inmunopatogenia de la diabetes Mellitus tipo 1 (23, 24), de la enzima de la polimerasa del virus de la hepatitis B con la proteína básica de la mielina, involucrada en la patogenia de la Esclerosis Múltiple (EM) (12), y fragmentos de la Yersinia y Clamidia involucrados en el desarrollo de PEP del tipo artritis reactivas (22).
Además, existen modelos experimentales en animales de desarrollo de autoinmunidad tras la inyección de antígenos virales o bacterianos como el desarrollo de Encefalomielitis Autoinmune Experimental Autoinmune (EAE) tras la infección por CMV, VHB y Hib (22, 23, 25); la participación de antígenos de virus de la familia herpes en el desarrollo de Síndrome de Sjögren (SS) (22, 25) y de la proteína gag del Retrovirus humano endógeno tipo 2 (HML-2) en AR (26).
Pese a esta evidencia, no basta la presencia de secuencias similares para gatillar autoinmunidad ya que esto sucede habitualmente en el proceso de presentación y reconocimiento antigénico en un individuo sano (21). Entonces lo que se plantea es que el MM requiere de la existencia de un “epítope relacionado con enfermedad”v(3), como lo ejemplifica la existencia del epítope compartido y el péptido artritogénico en la génesis de la AR y EAA antes comentado (7-10).
b. Activación por Espectador y Espectador Inocente (“innocent bystander”)Consiste en la activación de LT autoreactivos que no son específicos del agente infeccioso en cuestión, pero que responden a segundas señales generadas como consecuencia del daño tisular gatillado por el microorganismo (Activación por Espectador) (21) y al mismo tiempo involucra el daño secundario de tejidos propios que producto del ambiente proinflamatorio (citoquinas) y de la infección expresan en su superficie restos antigénicos del agente causal y moléculas coestimulatorias que facilitan su reconocimiento por LT autoreactivos que median el daño tisular (Espectador Inocente) (20, 21).
El mejor ejemplo de este mecanismo lo constituye el desarrollo de Diabetes tipo 1 en donde, tras la infección viral, las células profesionales presentadoras de antígenos (CPA), principalmente las células dendríticas maduras (CDm) expresan en su superficie moléculas coestimulatorias capaces de activar a LT autoreactivos de la periferia, quienes a su vez, reconocen los antígenos virales en la superficie de los islotes beta pancreáticos generando su destrucción por mecanismos dependientes de perforinas (27) y a la aparición del cuadro clínico característico (19, 21, 22).
Una situación similar ocurre en las infecciones por Mycoplasma pneumoniae en donde el daño tisular y la actividad del microorganismo daña la estructura de los eritrocitos, originando neoantígenos a partir de antígenos propios, ya que su conformación lineal o estructural es distinta a la original por lo que son reconocidos por LT, que al activarse, gatillan el desarrollo de anemia hemolítica autoinmune (22).
c. Infección PersistenteEste mecanismo implica el desarrollo de una RI mantenida en el tiempo producto de la persistencia en circulación de antígenos patógenos que expresados en la superficie de células sanas gatillan su reconocimiento y destrucción con el consecuente daño tisular del órgano involucrado (21).
Este mecanismo ha sido muy difícil de comprobar ya que si bien puede existir el antecedente epidemiológico de una infección previa, es muy bajo el número de cultivos y aislamientos que resulten positivos para el microorganismo involucrado (19, 20). No obstante hay evidencia de restos de los agentes involucrados identificados por biotecnología molecular que permiten apoyar la teoría (21).
Otro rol mencionado para la participación de infecciones en las enfermedades autoinmunes consiste en el desarrollo de reactivaciones. En este caso lo que se plantea es que dado que ya existen LT autoreactivos, el papel de los microorganismos consiste en amplificar la respuesta inmune mediante la inflamación del tejido blanco, la expresión de citoquinas proinflamatorias y el desarrollo de segundas señales coestimulatorias que exageran y perpetúan el proceso (21).
d. Activadores policlonales o Súper Antígenos (SSAA)Un SSAA es un producto proteico altamente conservado, habitualmente de origen bacteriano, que posee una enorme capacidad mitógena, que es la capacidad para estimular un gran número de linfocitos (potencialmente autorreactivos) en ausencia de su antígeno específico ya que lo realizan mediante un mecanismo independiente del complejo MHC y el receptor del LT o TCR al formar un complejo trimolecular con estas dos estructuras de forma directa pero sin procesamiento antigénico (18, 28). De este modo pueden generar una respuesta de LT generalizada e inespecífica (activación del 20 a 30% de LT circulantes) con liberación de citoquinas que llevan a cuadros sistémicos de fiebre, shock y muerte, como es el caso de las toxinas de Estreptococos y Estafilococos en los shock tóxicos o de EAI como se ha planteado en la patogénesis de la Enfermedad de Kawasaki (18, 28).
Pese a los anterior, ninguno de estos mecanismos en forma conjunta o independiente son capaces de gatillar por sí solos una enfermedad autoinmune. Esto porque las 3 situaciones antes mencionadas, son acontecimientos que habitualmente se desarrollan en el proceso de un cuadro infeccioso. La diferencia es que no todos los afectados, por ejemplo a una infección estreptocócica desarrollan necesariamente una enfermedad reumática. Aquí toma importancia el concepto del “terreno fértil” en donde los mecanismos relacionados con el medio ambiente deben llevarse a cabo en un terreno susceptible a desarrollar el proceso de autoinmunidad (3), terreno que como ya fue mencionado, dependerá de la susceptibilidad genética del individuo, de las características de sus sistema inmune y del ambiente que lo rodea.
Agentes InorgánicosSe ha descrito la participación de varias sustancias inorgánicas en la génesis y desarrollo de las EAI entre los que se incluyen hidrocarburos, pesticidas, fármacos, tinturas, suplementos dietarios, radiación ultravioleta y metales pesados (29, 30). Muchos de ellos son sólo asociaciones de frecuencia, pero algunos modelos experimentales han permitido demostrar su participación en la génesis o en el agravamiento de estas afecciones (30).
Un buen ejemplo lo constituye la exposición a metales pesados, como el mercurio (Hg) ya que existen modelos experimentales de EAI en roedores expuestos que demuestran como este estimula la apoptosis de las células cercanas a su lugar de inyección y estimula el clivaje de residuos proteicos generando neoantígenos los que son presentados como neoantígenos por las CPA (29). Además, altera las señales de transducción en células linfoides estimulando la expresión de marcadores de superficie celular de los LT relacionados con su activación o proliferación como CD 25, 44 y 71, así como de MHC tipo II en células presentadoras de antígenos (CPA), también genera la activación policlonal de LB con desarrollo de una hipergamaglobulinemia y generación de autoanticuerpos (29).
De este modo genera daño y al mismo tiempo activa al SI generando un círculo que lleva a la autoinmunidad y que clínicamente se manifiesta como el desarrollo de una glomerulonefritis membranosa, miocarditis y manifestaciones cutáneas del tipo enfermedad injerto versus huésped (29). Pese a lo anterior aún no hay una postura clara con respecto al uso y retiro de amalgamas dentales o a la aplicación de vacunas que poseen Hg como coadyuvante en pacientes con EAI (29).
HormonasEs conocido que del punto de vista epidemiológico, las EAI tienden a comprometer con más frecuencia a población del sexo femenino, por ejemplo se ha descrito que por cada 10 pacientes con LES sólo 1 es del sexo masculino y que por cada 4 pacientes con AR sólo uno es hombre (6, 31).
A su vez, el inicio de las EAI o la aparición de recidivas también se relaciona con cambios en el nivel de las llamadas hormonas sexuales, en donde la mayor población afectada involucra a mujeres en etapa fértil que tienden a debutar o desarrollar cuadros de reactivación clínica en etapas de cambios hormonales como lo son la menarquía, el embarazo, la lactancia y el hasta hoy controversial uso de anticonceptivos orales. Por otro lado, existe una tendencia a un menor número de casos de LES tras la menopausia, de modo que su incidencia pasa a ser muy similar a la del hombre (32).
En modelos murinos existe evidencia de la influencia de los estrógenos sobre las células del SI; por ejemplo el que linfocitos de pacientes con LES presentan mayor producción de autoanticuerpos tipo Anti DNA al ser tratados con estrógenos exógenos, mientras que el uso de testosterona redujo la producción (31, 32). De este modo, la respuesta a estrógenos puede exacerbar la enfermedad, mientras que la respuesta a andrógenos podría ser protectora (32).
Además, el uso de estrógenos bloquearía la apoptosis mediada por el receptor de antígenos del LB, mecanismo esencial para la eliminación de células B autorreactivas, ya que aumentan la producción de la proteína antiapoptótica Bcl-2 y estimulan la transcripción del factor de sobrevivencia de células B (BAFF) (17, 31, 32).
3Participación del sistema inmune en la autoinmunidadComo se menciono previamente, ciertas variaciones genéticas se traducen en la obtención de proteínas cuya funcionalidad puede alterar la RI del individuo. Sobre los principales componentes del SI involucrados en esta situación nos referiremos a continuación:
- a.
La Célula Dendrítica (CD): Es una CPA profesional y una de las responsables de la activación de LT autoreactivos ya que es la encargada de realizar la presentación de antígenos en los órganos linfoides secundarios donde se encuentran los LT (2). Es frecuente observar en algunas EAI como la tiroiditis o la AR una acumulación local de células inflamatorias y CD cuya disposición espacial semeja a la observada en un ganglio linfático (33).
En este caso, si la CD está madura (expresa en su superficie marcadores de activación celular), la presentación de antígenos ubicuos a un LT puede darse en un ambiente proinflamatorio y de este modo contribuir al quiebre de la tolerancia periférica (33).
El Factor activante de LB (BAFF): De la familia de TNF-α, estimula la sobrevivencia de los LB al inhibir la muerte celular programa por lo que ha sido señalada como la molécula responsable del rescate de LB autoreactivos anérgicos. Una excesiva producción de BAFF se ha relacionado con el desarrollo de EAI en modelos murinos del tipo LES o SS (2, 17, 34). FAS/FAS Ligando: La ausencia de alguno de ellos se traduce en el desarrollo poblaciones inmortales de LT y por lo tanto facilita el desarrollo de EAI y procesos linfoproliferativos (2). En el caso del CD95/Fas/Apo-1 así como de su ligando FasL, se han descrito tres mutaciones que han dado origen a tres cepas diferentes de ratones que tienden a desarrollar cuadros de linfoadenopatías masivas y una enfermedad tipo LES (35). Por otro lado, las células apoptóticas y los detritus celulares son consideradas las principales fuentes de neoantígenos en EAI como el LES (2). La combinación de una mayor tasa de apoptosis y un enlentecimiento en el clearance de estas podría incrementar la susceptibilidad al desarrollo de EAI (2, 35).
- b.
Receptores tipo Toll (TLR): Estos receptores de la respuesta inmune innata se caracterizan por reconocer patrones moleculares asociados a patógenos (PAMPs), por lo que su activación traduce eventos inflamatorios (2). Por ejemplo, el TLR tipo 2 puede activar CPA mediante la secreción de TNF-α, entonces, si su ligando son restos apoptóticos, por ejemplo de islotes beta pancreáticos en el contexto de una hepatitis viral, este evento podría ser un causal de quiebre de la tolerancia (15, 27). Lo mismo se ha descrito para el TLR tipo 4 que se activa por el lipopolisacáridos de las bacterias gram negativas involucradas en algunas enfermedades digestivas y respiratorias (15, 27).
- c.
Células Reguladoras: Son LT encargadas de regular y/o suprimir la actividad de otros LT cuya acción puede resultar dañina para el or ganismo (2). Entre sus funciones encontramos la prevención de EAI y alérgicas al ser las responsables de los mecanismos de anergia en la tolerancia periférica así como también de la tolerancia a antígenos alimentarios y materno-fetal. Su identificación es compleja ya que los marcadores que se han mencionado como característicos, son también marcadores de activación por lo que no son exclusivos de esta población (CD 25, CTLA-4, CD 127, LAG-3 y FoxP3) (33). Se ha descrito un menor número y/o una menor actividad en esta población en los pacientes con EAI tipo LES y SS (33).
- d.
Citoquinas: Son mediadores y efectores de la RI, cuyas funciones redundantes y pleiotrópicas pueden influir en el desarrollo y perpetuación de cuadros inflamatorios (2, 14). Por ejemplo, es conocido el rol de las citoquinas proinflamatorias como IL-1, IL-6 y TNF-α en amplificar y mantener la RI y se ha logrado identificar a la IL-6 como una de la principales citoquinas involucradas en la patogenia del Still (14). También se la mencionado el rol de IL-8 como quimioatractante y responsable del influjo de células inflamatorias al tejido lesionado como un factor perpetuador de la inflamación sinovial en los pacientes con AR (36). Existen reportes de un desbalance en la regulación normal de la RI en casi todas las EAI y se ha empleado la medición de sus niveles pueden un indicador de la actividad de la EAI, (principalmente intra articular, pero no necesariamente con la inflamación sistémica) (15, 36).
Además, pueden mediar la expresión aberrante de MHC clase II en células no inmunes, transformándolas en presentadoras involuntarias capaces de activar a LT autoreactivos en un ambiente proinflamatorio, lo que se ha descrito para tirocitos en la tiroiditis autoinmune y en células pancreáticas de la DM tipo 1 (15, 36). Finalmente, se han desarrollado nuevas herramientas terapéuticas que involucran el bloqueo de las acciones de las ILs como una forma de controlar los sus efectos deletéreos. El ejemplo más clásico lo conforman los llamados fármacos “anti TNF” que mediante proteínas de fusión o anticuerpos monoclonales son capaces de limitar la acción del TNF-α (34).
Las células Th 17 corresponden a una población de LT Helper descritas recientemente y definidas como tal por su producción de IL-17. Pese a que no está claro su mecanismo de generación en humanos varios grupos han demostrado que la IL-1β, 6, 23 y el factor de crecimiento transformante beta (TGF-β) influirían en la diferenciación de esta población a partir de células naive (37-39). Su aparición deja obsoleto el antiguo paradigma de respuesta Th1/Th2 (37) y su importancia radica en que la IL-17 jugaría un rol crítico en la patogenia de las EAI al ser considerada una citoquina proinflamatoria dentro de cuyas funciones destacan la producción de citoquinas y moléculas proinflamatorias como IL-6, prostaglandina E2 y óxido nítrico, quimioatractante para monocitos y neutrófilos al aumentar la expresión de moléculas coestimulatorias y de adhesión celular, acción sinérgica con otras citoquinas proinflamatorias (IL-1β, TNF-α e IFN-γ) y otras aún es estudio pero con la finalidad común de amplificar la respuesta inflamatoria (38, 39). Existen además modelos murinos de AR que demuestran la presencia de células Th 17 en la sinovial de las articulaciones comprometidas(38, 39). La participación y función de esta población en la patogenia de las EAI es aún materia de debate, pero ya se visualizan como futura herramienta terapéutica.
Una vez que hemos revisado cada uno de los actores involucrados en el desarrollo de la autoinmunidad es más fácil entender cómo cada uno de ellos debe entrelazarse con el siguiente para dar origen a una enfermedad autoinmune. Revisaremos algunos modelos de autoinmunidad que involucran a los actores ya mencionados:
Modelo del antígeno críptico y la ignoranciaEn un ambiente proinflamatorio, por ejemplo producto de una infección, isquemia o traumatismo, donde confabulan los mecanismos ya descritos, el daño tisular de los órganos blancos puede exponer al SI antígenos que se encontraban de una forma críptica o secuestrada (esperma y proteínas intraoculares), lo que genera un reconocimiento antigénico y una RI contra ellos ya que estos antígenos habían sido “ignorados” por el SI pero no se ha desarrollado tolerancia hacia ellos. Este evento explica el desarrollo de enfermedades como la oftalmopatía simpática refleja del ojo sano tras un traumatismo del ojo contralateral o el desarrollo de una orquitis linfocitaria tras la realización de una vasectomía.
Modelo del speading o propagación del epítopeEl ataque autoinmune genera daño tisular donde inevitablemente hay formación y liberación de nuevos antígenos que potencialmente serán reconocidos por otros LT autoreactivos exacerbando el daño y propagando la enfermedad (2).
Este concepto se conoce como propagación del epítope y permite explicar por que cuando se produce una EAI esta tiende a ser crónica y progresiva. Al mismo tiempo permite explicar cómo una enfermedad órgano específica como las tiroiditis autoinmunes pueden ser el factor inicial de una enfermedad sistémica como el LES, cómo el desarrollo de anticuerpos que un paciente posee al inicio de una enfermedad va cambiando y ampliándose en el tiempo y cómo un blanco antigénico inicial puede ampliarse a otros (2). Ejemplos de esto es lo que sucede en el síndrome de Good Pasture donde el antígeno inicial es la cadena alfa 3 del colágeno tipo 4 presente en la membrana basal glomerular, pero en la medida en que el daño progresa se desarrolla reactividad cruzada contra otros antígenos del colágeno, como los existentes en la membrana alveolar, explicando la aparición de nuevas especificidades antigénicas y daño de otros órganos blancos (2). Lo mismo sucede con las enfermedades ampollares como el pénfigo en donde el antígeno inicial corresponde a la desmogleina 1 (proteína constituyente de los desmosomas) que por su ubicación es responsable de la aparición de bulas mucosas, pero a medida que aumenta el daño inmune se expresan otras desmogleinas como la tipo 3 generando la aparición de nuevos autoanticuerpos y de lesiones epidérmicas, por ser la desmogleina 3 uno de los principales constituyentes de los desmosomas de la epidermis (Teoría de descompensación de la desmogleina) (40).
ConclusiónTeniendo en consideración la complejidad de los mecanismos de tolerancia, la importancia de la susceptibilidad genética y la participación de actores externos es casi imposible definir un modelo único de autoinmunidad. Por esto es que se plantea que el resultado de las EAI, a groso modo, depende de la ocurrencia de tres pasos a saber: Un evento iniciador (infección, injuria u otro), un terreno genéticamente susceptible y un sistema inmune desregulado.
Los autores declaran no tener conflictos de interés, con relación a este artículo.