






Las interacciones farmacológicas representan un problema mayor en el manejo de los pacientes trasplantados. La comprensión de los distintos pasos del metabolismo de estos fármacos permite anticipar y prevenir complicaciones derivadas de su uso. Cada nuevo medicamento introducido en la terapia de estos pacientes debe ser acompañado de una revisión de las interacciones con los inmunosupresores y otros fármacos prescritos.
Drug interactions are a major problem in the management of transplant patients. Understanding the various steps in the metabolism of these drugs allows us to anticipate and prevent complications arising from their use. Each new drug introduced in the therapy of these patients should be followed by a review of interactions with immunosuppressive agents and other drugs prescribed.
El paciente trasplantado se ve sometido a una serie de problemas médicos, destacando entre estos los efectos adversos de los fármacos inmunosupresores. Estos medicamentos causan efectos adversos conocidos, pero además pueden interaccionar entre ellos y con otra serie de fármacos que habitualmente recibe este tipo de paciente. La necesidad de tratar problemas como la hipertensión arterial, diabetes, gota, infecciones, dislipidemia, osteoporosis, cáncer y la aparición continua de nuevos fármacos enfrenta al paciente y equipo médico ante el riesgo de interacciones cada vez más frecuentes. En el paciente trasplantado están presentes gran parte de los riesgos clásicos de interacciones farmacológicas, como son polifarmacia, edad avanzada, fármacos con umbral terapéutico estrecho o que requieren un monitoreo estricto.
En este artículo revisaremos los mecanismos involucrados en este tipo de interacciones y las pautas para evitar los riesgos potenciales.
Principios generalesLas interacciones entre fármacos pueden ser farmacocinéticas o farmacodinámicas. La interacción farmacocinética puede producirse en la absorción, distribución, metabolismo o eliminación de una droga. La interacción farmacodinámica ocurre cuando una droga disminuye o aumenta la acción de otra o altera su toxicidad. Las interacciones farmacocinéticas pueden producirse a nivel de tracto gastrointestinal, circulación enterohepática, sitios de unión a tejidos o proteínas, excreción biliar, enzimas metabólicas de drogas y sistema de transporte.
Luego de la administración oral de un inmunosupresor varios factores afectan la absorción oral como: la cantidad que llega al intestino (que a su vez se afecta por el pH gástrico, presencia de alimento en estómago o su vaciamiento), absorción, metabolismo intestinal y el primer paso hepático. La absorción se produce principalmente en intestino delgado y los agentes procinéticos aceleran la absorción de inmunosupresores como los anticalcineurínicos.
Entre los factores principales destaca el hecho que muchos inmunosupresores son metabolizados en el hígado a través del sistema enzimático conocido como citocromo P450 (CYP450). Este sistema se ubica también en intestino, dónde se acompaña de una proteína transportadora de drogas llamada P-glicoproteína (P-gp), que tiene como función contra-transportar los fármacos hacia el lumen intestinal. El CYP450 pertenece a una superfamilia de enzimas de tipo oxigenasas. Este sistema participa en la primera fase (fase I) del metabolismo hepático de una droga (Figura 1). Se ubican en el retículo endoplásmico liso y catalizan la biotransformación de las drogas en compuestos más hidrosolubles. En general las enzimas CYP producen metabolitos con menor acción biológica, sin embargo existen múltiples excepciones que se refieren a la generación de productos activos con efectos tóxicos o carcinogénicos. Se conocen 57 tipos de formas de CYP en los seres humanos y la mayoría se expresa en el hígado. Se agrupan por familias (CYP 1-10) y subfamilias. Las familias de CYP 4 a 10 están encargadas del metabolismo de compuestos endógenos y no son inducibles por sustancias exógenas (drogas). Tres familias (CYP1, CYP 2 y CYP3) son las más importantes en el metabolismo hepático de sustancias exógenas y drogas. Más del 50% de los fármacos utilizados en la práctica clínica son metabolizados por la subfamilia CYP3A. Entre estos destacan gran parte de los inmunosupresores conocidos y muchos medicamentos de uso común. En la población general este sistema presenta un polimorfismo genético importante. Estas alteraciones genéticas pueden determinar un menor metabolismo, ausencia de metabolismo o aumento de actividad metabólica sobre una droga en particular.

La segunda fase del metabolismo hepático (fase 2) es la conjugación de los metabolitos producidos en la fase I con moléculas hidrosolubles (ácido glucurónico, sulfato, acetato…). En general, esta reacción crea sustancias inactivas, no tóxicas y fáciles de excretar.
La inhibición del sistema CYP450 origina un menor metabolismo de drogas, que determina una mayor exposición al fármaco y por tanto mayor riesgo de toxicidad específica. Al contrario, la inducción del sistema causa una mayor cantidad de enzimas ya sea por mayor síntesis o menor degradación. Es así que 2 o más fármacos interactúan a este nivel y pueden cambiar el metabolismo del otro y por tanto su nivel plasmático y por tanto sus efectos terapéuticos y reacciones adversas (Figura 2). La inhibición de estas enzimas se produce rápidamente luego de administrar una droga inhibidora, pero la inducción enzimática es un proceso más lento que puede tardar 2 semanas.
Monitoreo terapéutico
La medición de los niveles de los fármacos inmunosupresores es una práctica habitual en clínica. Se utilizan los niveles valle, los niveles pico (Cmax), y la estimación del área bajo la curva (AUC). La medición de estos niveles debe realizarse siempre que exista riesgo de interacción. Su frecuencia u oportunidad dependerá de la importancia de la interacción, sus riesgos clínicos y su tiempo de aparición. En general los pacientes trasplantados son más susceptibles a las interacciones durante los primeros meses de trasplante o si han presentado episodios de rechazo agudo.
A continuación revisaremos las interacciones específicas de los inmunosupresores habituales.
Interacción de Ciclosporina y Tacrolimus (anticalcineurínicos)Los fármacos Ciclosporina (CSA) y Tacrolimus (TAC) son metabolizados en hígado por el sistema enzimático CYP450 3A. La CSA es extensamente metabolizada en hígado por esta enzima y menos del 0.1% es excretada como tal en orina. Se identifican cerca de 25 metabolitos de CSA en sangre, orina, bilis y heces. Estos metabolitos son menos activos y tóxicos que la droga madre. La CSA interacciona en forma importante con varios fármacos de uso común y productos naturales. Cualquier sustancia que afecte la acción de CYP3A puede alterar la concentración plasmática de CSA. Los inhibidores de CYP3A reducirán el metabolismo de CSA incrementando el riesgo de toxicidad. Considerando estas interacciones se utiliza la asociación con Ketoconazol y Diltiazem para disminuir costos al reducir la dosis de CSA en un 80% y 50% respectivamente en los pacientes trasplantados. Otro ejemplo es el jugo de pomelo que debe evitarse en pacientes trasplantados porque puede aumentar el AUC de CSA hasta en 200%. Al contrario los estimuladores de CYP3A causan una caída en los niveles plasmáticos de CSA y con esto aumentan el riesgo de rechazo. Un ejemplo son los anticonvulsivantes Fenitoína y Fenobarbital que reducen en 50% la biodisponibilidad de CSA. En la Tabla 1 se detallan los fármacos habituales que pueden interactuar con CSA y TAC. A su vez tanto CSA como TAC reducen la actividad de CYP3A en un 30%, lo que afecta el metabolismo de otras drogas que utilizan este sistema.
Fármacos que aumentan o disminuyen los niveles de ciclosporina (CSA) y tacrolimus (TAC)
| Aumento de los niveles de CSA o TAC | Baja de los niveles de CSA o TAC |
|---|---|
| Diltiazem | Rifamopicina |
| Verapamilo | Carbamazepina |
| Fluconazol | Fenobarbital |
| Itraconazol | Fenitoína |
| Ketoconazol | Octreotide |
| Claritromicina | Ticlopidina |
| Eritromicina | Orlistat |
| Lansoprazol | |
| Alopurinol | |
| Bromocriptina | |
| Colchicina | |
| Amiodarona |
Todos los azoles inhiben en con distinta potencia el CYP3A4, siendo el Ketoconazol el más potente seguido de Itraconazol, Posaconazol, Voriconazol y Fluconazol en orden decreciente. El Ketoconazol aumenta el AUC de CSA en 3 veces y se ha utilizado para reducir los costos del uso de CSA. El Voriconazol, agente de amplio espectro, eleva en 1.7 veces el AUC de CSA y se recomienda reducir la dosis de esta última en 50% al combinarse. Similar efecto tiene sobre el AUC de TAC, recomendándose reducciones de dosis de 60 a 70%. En caso de uso de Posaconazol se recomienda una reducción de dosis de 25% para CSA y de 66% para TAC.
Una nueva clase de antifúngicos de amplio espectro son las equinocandinas, representadas por la Caspofungina. Estos fármacos no afectan el sistema CYP450. Sin embargo en el uso concomitante con CSA se ha reportado elevación de transaminasas en algunos pacientes. Otros reportes en trasplante hepático con CSA o TAC no muestran hepatotoxicidad al utilizar Caspofungina. Se recomienda la monitorización de pruebas hepáticas al utilizar esta nueva clase de antifúngicos junto con anticalcineurinicos.
La anfotericina-B se utiliza en las micosis invasoras y es nefrotóxica. Esta toxicidad se ve aumentada en trasplantados por la disfunción renal pre-existente y el uso concomitante de los anticalcineurínicos. Si no hay alternativas a su uso, deben preferirse las formas liposomales de anfotericina e hidratar adecuadamente al paciente antes y después de administrarla.
La Rifampicina es un potente inductor de CYP3A4 y es capaz de reducir la concentración valle de CSA en 73%. Si se utilizan en forma combinada debe aumentarse la dosis de CSA en 2.5 a 3 veces. Con TAC ocurre algo similar y se reportan incrementos de 10 veces en su dosis para mantener niveles adecuados.
Interacción con antihipertensivosLos bloqueadores de los canales de calcio no dihidropiridínicos Verapamilo y Diltiazem son potentes inhibidores del CYP3A y P-gp, por lo que incrementan el nivel de CSA y TAC entre 1.5 a 6 veces. Se recomienda en caso de uso con CSA o TAC, reducir la dosis de estos entre 25 a 75%. Esta interacción es menor para los bloqueadores de los canales de calcio dihidropiridínicos, como Amlodipino, Felodipino y Nicardipino. Sin embargo el efecto parece ser mayor para Nifedipino.
Los beta-bloqueadores son seguros y bien tolerados en trasplante. Solo se ha reportado un incremento en los niveles valle de CSA al iniciar Carvedilol, al parecer por inhibición de la P-gp.
Interacción con HipolipemiantesLos fármacos de la familia de las estatinas son substratos metabólicos de CYP3A4, por lo que el uso concomitante con CSA o TAC favorece la toxicidad muscular de estos fármacos. Todas las estatinas presentan riesgo de rabdomiolisis al utilizarlas junto con CSA, salvo Fluvastatina metabolizada a través de CYP2C9. En cambio la Pravastatina y Rosuvastatina tienen solo un metabolismo mínimo por el sistema CYP. El riesgo aumenta en relación a la dosis de estatina utilizada. Por lo que se recomienda utilizarlas en la menor dosis posible y preferir aquellas con metabolismo diferente al sistema CYP. En un estudio reciente el TAC no mostró interacciones significativas con Atorvastatina, sin embargo se desconoce si esta ausencia de interacciones se aplica a otras estatinas. En general, cuando se administra un anticalcineurínico junto con una estatina el paciente debe ser seguido de cerca en busca de síntomas o signos de miopatía o rabdomiolisis.
Los fibratos son metabolizados por CYP3A4 y excretados por vía renal. Los estudios sugieren una reducción del AUC de CSA con estos fármacos. La combinación entre fibratos, estatinas y CSA incrementa máximamente el riesgo de toxicidad muscular.
Se ha reportado que la CSA aumenta la concentración plasmática de Ezetimibe en 12 veces.
Interacción con anti-plaquetariosLa Ticlopidina reduce la concentración de CSA a través de la inducción del CYP3A. No existen datos con TAC. Por otra parte CSA y TAC pueden reducir la concentración del metabolito activo del Clopidogrel, disminuyendo el efecto anti-plaquetario.
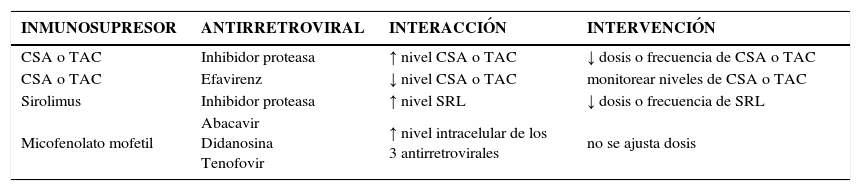
Interacción con terapia antirretroviralGracias a la exitosa terapia antirretroviral los pacientes VIH han ingresado a los programas de trasplante, sin embargo con esto se han evidenciado las interacciones de estas terapias con los inmunosupresores. Los inhibidores de proteasa causan una significativa inhibición de CYP3A4 y Pgp, lo que se traduce en un incremento en el nivel plasmático de sus sustratos. Cuando se administran junto con los anticalcineurinicos se debe disminuir la dosis de CSa o TAC en forma significativa. En trasplante renal la combinación Lopinavir/Ritonavir incrementa en 10 veces la vida media de TAC y la combinación Nelfinavir/Indinavir obliga a reducir en 85% la dosis de CSA. En trasplante hepático la introducción de Nelfinavir obliga a reducir la dosis de TAC entre 16 a 70 veces. Por el contrario los inhibidores de la transcriptasa reversa, como el Efavirenz y la Nevirapina, estimulan el metabolismo de sustratos del CYP3A4 como CSA. Cualquier cambio en la terapia antirretroviral debe ser seguido de una vigilancia en los niveles de inmunosupresión para evitar ya sea toxicidad o mayor riesgo de rechazo. En la tabla 2 se señalan interacciones importantes entre inmunosupresores y antirretrovirales (Tabla 2).
Interacciones de la terapia antirretroviral con inmunosupresores
| INMUNOSUPRESOR | ANTIRRETROVIRAL | INTERACCIÓN | INTERVENCIÓN |
|---|---|---|---|
| CSA o TAC | Inhibidor proteasa | ↑ nivel CSA o TAC | ↓ dosis o frecuencia de CSA o TAC |
| CSA o TAC | Efavirenz | ↓ nivel CSA o TAC | monitorear niveles de CSA o TAC |
| Sirolimus | Inhibidor proteasa | ↑ nivel SRL | ↓ dosis o frecuencia de SRL |
| Micofenolato mofetil | Abacavir Didanosina Tenofovir | ↑ nivel intracelular de los 3 antirretrovirales | no se ajusta dosis |
La depresión es frecuente en los pacientes trasplantados o en lista de espera. En general se utilizan los inhibidores selectivos de la recaptación de serotonina. Entre estos la Nefazodona y la Fluvoxamina son potentes inhibidores del CYP3A4 y pueden elevar los niveles de CSA hasta 10 veces. La Fluoxetina, Sertralina, Mirtazapina y Paroxetina tienen un efecto inhibitorio débil sobre el CYP3A4. Una alternativa terapéutica es la Venlafaxina, que es metabolizada por otra isoenzima, el CYP2D6.
Fuera de las interacciones citadas se debe señalar que otros psicofármacos pueden ver aumentada su toxicidad en caso de deterioro de función renal por los anticalcineurínicos. Este es el caso del Haloperidol, Quetiapina u Olanzapina que pueden prolongar el intervalo QT.
Interacción con antiepilépticosEl uso concomitante de CSA o TAC con Fenitoína reduce los niveles de estos inmunosupresores y puede elevar la concentración plasmática del antiepiléptico. Este último efecto se debe a la gran unión a proteínas plasmáticas de CSA y TAC y el consecuente desplazamiento de la Fenitoína de su proteína transportadora. La Carbamazepina y Oxcarbazepina son potentes estimuladores del metabolismo de CSA y TAC y pueden reducir sus niveles hasta 4 veces. Este efecto puede durar hasta 4 meses post suspensión de la droga. Fármacos alternativos que no inhiben la CYP3A4 son el Ácid Valproico, Gabapentina, Lamotrigina y Vigabatrina. Otra alternativa anticonvulsivante es el Levetiracetam que no es metabolizado en hígado y por tanto su uso es seguro en trasplante.
Interacción con anti-gotososLa CSA puede producir una acumulación de Colchicina y sus metabolitos en los tejidos. Esto causa un cuadro clínico de náuseas, vómitos, mialgias, debilidad muscular, ictericia y parestesias. Se recomienda el uso de Colchicina por tiempo breve, a dosis bajas y suspender si aparecen síntomas.
Inhibidores de mTORInteracción de Sirolimus y EverolimusSirolimus o Rapamicina (SIR) tiene una biodisponibilidad baja (14% de la dosis oral) dado su extensa metabolización en intestino e hígado por CYP3A4 y su co-transporte intestinal por P-gp. Uno de sus metabolitos es el Everolimus (EVER), fármaco activo. Al presentar un metabolismo por CYP3A4 se ve afectado por las mismas drogas que afectan a CSA y TAC. A su vez SIR reduce la actividad de CYP3A en 38% y su uso combinado con CSA la reduce en 55%, aumentando la toxicidad renal de Inhibidores de la calcineurina como CSA y TAC.
Interacción con fármacos habitualesLos antifúngicos azoles (Ketoconazol, Voriconazol, Fluconazol) tiene un efecto potente en cuanto a incrementar la AUC y la Cmáx de SIR y EVER. Los bloqueadores de los canales de calcio tienen un efecto moderado sobre SIR y EVER. Las estatinas en cambio tienen un efecto débil.
Interacción con Ciclosporina y TacrolimusLa administración simultánea de SIR y CSA produce un aumento significativo de los niveles pico y valle del SIR. En cambio si la dosis de SIR era administrada 4 horas después de la de CSA se producía un incremento 3 veces menor. Además del problema de interacción farmacológica, la combinación CSA-SIR aumenta la hipertrigliceridemia, la nefrotoxicidad y el riesgo de síndrome hemolítico urémico. La fórmula modificada de la CSA incrementa la Cmáx y la AUC de EVER significativamente, por mecanismos no claros.
Aunque SIR y TAC tienen un mecanismo de acción diferente, comparten la misma proteína transportadora (FKBP-12). A pesar de esto, los estudios no muestran inhibición competitiva. A diferencia de CSA, la administración conjunto de TAC y SIR no altera sus niveles plasmáticos. Sin embargo, se ha reportado en receptores de trasplante renal que utilizaron la combinación de TAC-SIR una nefropatía similar a la de las cadenas livianas del mieloma múltiple.
Interacción con MicofenolatoEl uso conjunto de SIR y Micofenolato Mofetil (MMF) eleva significativamente la AUC de MMF, a diferencia del uso de CSA con MMF. Se recomienda una reducción en la dosis de MMF al 50% al combinarlo con SIR.
Agentes anti proliferativosInteracción de MicofenolatoEl MMF es rápidamente absorbido en tubo digestivo y transformado por esterasas hepáticas en ácido micofenólico. Este último es inactivado por glucuronidación y eliminado por riñón. Entre 8–12 horas post dosis se observa un segundo pico en el nivel plasmático causado por la recirculación entero-hepática. El MMF no tiene un efecto significativo sobre la actividad de CYP3A.
La absorción de MMF se ve afectada por la toma simultánea de antiácidos o hierro oral, por lo que se debe separar su ingesta por 2 a 4 horas. La Colestiramina es capaz de reducir los niveles de MMF al interferir en su circulación entero-hepática, por esto se recomienda no administrar simultáneamente. Los niveles de MMF son reducidos por CSA al interferir en la recirculación entero-hepática, un efecto que no tiene TAC ni los inhibidores de mTOR. Por esto se recomienda vigilar los efectos tóxicos y reducir la dosis de MMF luego de cambiar un paciente de CSA a TAC o inhibidores de mTOR.
Especial mención requiere la asociación de inhibidores de mTOR y MMF en relación a anemia y aumento del tránsito intestinal que puede llegar a ser motivo para el retiro de una de ellas.
Interacción de AzatioprinaLa Azatioprina (AZA) es una pro-droga, siendo convertida rápidamente a 6-mercaptopurina. Esta última puede seguir distintas vías y dar origen a otros metabolitos, entre ellos destaca la 6-tioguanina, principal causante de la toxicidad medular. Un porcentaje de la población (11%), por causas genéticas, presenta una actividad reducida de las enzimas necesarias para evitar la acumulación de este metabolito tóxico.
El Alopurinol y su metabolito activo inhiben a la enzima xantino-oxidasa, incrementando el nivel de 6-mercaptopurina. Se reportan múltiples de casos de leucopenia, anemia y trombocitopenia al combinar Alopurinol y AZA. Se recomienda evitar la asociación o al menos reducir las dosis de ambos en 70 a 80%. La triple asociación con inhibidores de la ECA (IECA), en algunos pacientes incrementa severamente el efecto de supresión medular, por lo que se recomienda cautela o evitar su indicación cambiando AZA por Micofenolato de ser necesario.
La AZA en dosis altas (> 100mg/día) puede causar resistencia al efecto anticoagulante de la Warfarina.
ConclusionesEl predecir una interacción farmacológica en un trasplantado es difícil. Estos pacientes toman una gran cantidad de fármacos inmunosupresores y no inmunosupresores, con riesgos potenciales de efectos adversos graves. Existe una relación bidireccional entre el sistema citocromo P450 y los distintos fármacos utilizados. El comprender el funcionamiento del sistema CYP ayuda a predecir posibles interacciones entre fármacos. Las interacciones entre drogas siguen siendo un problema clínico importante que afecta a los receptores de trasplante. Por esto deben ser constantemente supervisadas cuando se añade o se elimina algún medicamento a estos pacientes.
Los autores declaran no tener conflictos de interés, en relación a este artículo.