







Las atrofias musculares espinales no-5q son un conjunto de entidades hereditarias, clínica y genéticamente heterogéneas secundarias a compromiso de las células del asta anterior de la médula. No están asociadas a deleción del gen de sobrevida de la motoneurona (SMN1) responsable de la forma más conocida y clásica de atrofia muscular espinal. Identificar estos cuadros por sus características específicas y diferenciarlos de la atrofia muscular espinal clásica, es una necesidad imperiosa, ahora más que nunca, dada la existencia de un reciente tratamiento aprobado para la atrofia clásica y el conocimiento de que algunas formas no clásicas son susceptibles de mejorar con tratamientos específicos. Este es el caso de aquellas secundarias al déficit del transportador de riboflavina, cuyo diagnóstico y tratamiento oportuno permite salvar la vida del paciente.
En esta revisión abordaremos la clasificación clínica actual de estos cuadros con sus más de 18 genes involucrados, las formas de presentación, evolución diagnóstica y tratamientos de las más significativas.
Non-5q spinal muscular atrophies are different hereditary anterior horn cell diseases, clinical and genetically heterogeneous, not associated with a homozygous deletion of the motor neuron survival gene (SMN1) underlying the most known and classic form of spinal muscular atrophy. Identify these entities by their specific clinical features and be able to differentiate them from classical spinal muscular atrophy is an urgent need. Now more than ever, given the newly approved drug, to treat classic spinal muscular atrophy and the emerging knowledge that some non-classical forms also have a specific treatment. Such is the case for the deficiency of riboflavin transporters where timely diagnosis and treatment may allow saving the life of patients.
In this review, we will address the current clinical classification of these diseases with their more than 18 genes involved, the forms of presentation, diagnostic, evolution and treatments of some of these.
Atrofia muscular espinal (AME) es el nombre usado para describir un grupo de desórdenes genéticos caracterizados por debilidad y atrofia muscular progresiva, secundarios al compromiso de la segunda motoneurona a nivel espinal y bulbar inferior1. La severidad clínica y heterogeneidad genética es amplia y se extiende desde fenotipos de inicio temprano, con debilidad proximal simétrica de extremidades, asociada a compromiso respiratorio con muerte prematura, hasta una afectación más benigna restringida a debilidad distal no progresiva de las extremidades inferiores1–4. Los pacientes con AME se pueden subdividir mediante análisis genéticos en dos grupos:
- (1)
AME clásica, secundaria a la deleción o mutación del gen de supervivencia de la motoneurona (Survival Motor Neuron, SMN1) ubicado en el cromosoma 5q11.2-q13.3. En aproximadamente un 95% a 98% de los pacientes se produce por deleción homocigota del exón 7. En los casos restantes, se puede presentar mutación puntual en un alelo y deleción del exón 7 en el otro (heterocigoto compuesto)1.
- (2)
AME no clásica, secundaria a mutaciones en numerosos otros genes, también conocidas como AME no-5q.
La AME clásica es la más frecuente, representa el 80-90% de los trastornos hereditarios de la motoneurona, con una incidencia es 1 en 6000-10000 nacidos vivos, es la causa genética más común de mortalidad infantil en todo el mundo5–7. En tanto, en AME no-5q, la frecuencia es difícil de precisar debido a la gran variabilidad de genes involucrados y al continuo incremento de nuevos genes responsables, alcanzando en algunas series hasta un 4% de todas las AME8. Suelen clasificarse según el tipo de herencia y patrón predominante de debilidad en proximal, distal o bulbar. Cuando la afección clínica predominante afecta segmentos distales de las extremidades también se les denomina neuropatías motoras hereditarias distales (dHMN) o atrofias musculares espinales distales (dSMA). Si bien, la presentación clínica de algunos tipos de AME no-5q puede ser similar a las AME clásica, el estudio complementario, el tratamiento y el asesoramiento genético son claramente diferentes1. Muchas de estas formas de atrofias musculares espinales se acompañan de otros síntomas o compromisos no presentes en la forma clásica de la enfermedad por lo que algunos investigadores clínicos también las han llamado “SMA Plus Syndromes”4; esto genera que el espectro fenotípico de AME no-5q se superponga con otras entidades como neuropatía sensitiva y motora hereditaria, esclerosis lateral amiotrófica y la paraparesia espástica hereditaria9 (figura 1).
 Distribución habitual de atrofias musculares espinales distales con predominio de amiotrofia y debilidad en extremidades inferiores. (C) Pie equino y amiotrofia distal bajo la rodilla. (D) Lactante con contracturas y deformidad de manos y pies. (E) Amiotrofia musculatura intrínseca de la mano, eminencia tenar e hipotenar. (F) Escapula alada. (G) Atrofia y fasciculaciones linguales. Fotos autorizadas por padres y pacientes.")
Manifestaciones clínicas de enfermedades con compromiso de segunda motoneurona
(A y B) Distribución habitual de atrofias musculares espinales distales con predominio de amiotrofia y debilidad en extremidades inferiores. (C) Pie equino y amiotrofia distal bajo la rodilla. (D) Lactante con contracturas y deformidad de manos y pies. (E) Amiotrofia musculatura intrínseca de la mano, eminencia tenar e hipotenar. (F) Escapula alada. (G) Atrofia y fasciculaciones linguales.
Fotos autorizadas por padres y pacientes.
Los mecanismos patogénicos subyacentes al daño de la motoneurona son variados y no completamente comprendidos, incluyen la alteración de diversos procesos celulares como la regulación del control de la calidad de la autofagia, la dinámica del citoesqueleto y el metabolismo del ARN. Otros mecanismos adicionales incluyen anomalías estructurales y funcionales de las mitocondrias, estrés oxidativo mediado por radicales libres, transporte molecular por canalización de cationes (TRPV4), captación de vitaminas (SLC52A3 y SLC52A2), transporte nuclear (GLE1), metabolismo lipídico (ASAH1) y transporte axonal (BICD2 y DYNC1H1)4.
Las nuevas técnicas de estudios genéticos moleculares, especialmente la secuenciación de nueva generación con paneles de genes específicos, secuenciación exómica y genómica han revolucionado las posibilidades de acceso a diagnósticos de precisión y con ellos los clásicos algoritmos de estudio de los pacientes, haciendo más relevante una cuidadosa descripción y estudio fenotípico que facilite la precisión en la interpretación de los hallazgos moleculares10. Así mismo, debemos resaltar la aparición de nuevas terapias que comienzan a cambiar la historia natural de enfermedades neurodegenerativas como la AME clásica, así como del incremento en la comprensión de los mecanismos patogénicos subyacentes a la degeneración de la motoneurona, que posibilita los desarrollos terapéuticos en estas enfermedades11–13. El objetivo de este artículo es dar una perspectiva general de la atrofia muscular espinal, revisando las características clínicas y genéticas, particularmente, de los diagnósticos diferenciales que se encuentran agrupados bajo el concepto de AME no-5q, proponiendo puntos claves para la aproximación diagnóstica y terapéutica.
PRESENTACIÓN CLÍNICACuando enfrentamos pacientes con enfermedades que comprometen a las motoneuronas inferiores, la principal manifestación clínica es la debilidad muscular. Esta debilidad puede ser de predominio proximal o distal, comprometer en forma simétrica o asimétrica las extremidades, ser de predominio en extremidades inferiores o superiores o a veces generalizada y en ocasiones también afectar la musculatura respiratoria: intercostales, diafragma y acompañarse de insuficiencia respiratoria. En otras oportunidades también aparecen trastornos de la deglución y de la voz si el daño alcanza las motoneuronas bulbares14. La disminución o ausencia de reflejos osteotendíneos, la existencia de fasciculaciones musculares o linguales, temblor fino de manos o poliminimioclonus nos advierten que debemos orientar nuestro estudio hacia la certificación de un compromiso de segunda motoneurona (figura 1)3. La electromiografía y velocidad de conducción nerviosa certifican la presencia de signos de denervación activa como fibrilaciones y ondas agudas positivas y/o reinervación crónica con potenciales de unidad motora de duración y tamaño aumentado, polifásicos y reclutamiento reducido. Los estudios de conducción motora señalan habitualmente velocidades de conducción normales con amplitudes disminuidas por la pérdida axonal; mientras que, el estudio de conducción sensitiva suele ser normal15. La creatinquinasa sérica está normal o levemente aumentada en estos pacientes, y en ocasiones la necesidad de orientar el diagnóstico en las formas más complejas hace necesario la realización de una biopsia muscular que mostrará el característico aspecto de atrofia neurogénica o “agrupamiento de fibras”16.
Otro aspecto importante que considerar, es la edad de inicio de los síntomas; congénito, de inicio infantil precoz, tardío juvenil, o edad adulta y precisar el patrón evolutivo; considerando que existen cuadros rápidamente progresivos, otros de progresión lenta e incluso algunos estacionarios. Los siguientes elementos necesarios de precisar son los eventuales compromisos de otros sistemas: auditivo, visual, cerebeloso, cortical, la existencia de epilepsia, discapacidad intelectual, ginecomastia, alteraciones de la voz, endocrinológicas, gastrointestinales, urinarias, artrogriposis, fracturas congénitas y extender nuestra investigación con exámenes que identifiquen estos eventuales compromisos: resonancia de cerebro y médula, electroencefalograma, electrocardiograma, ecocardiograma, estudios audiológicos, entre otros4 (Figura 1).
Este abordaje inicial del paciente, junto con una detallada historia familiar, presencia de otros miembros afectados, y el conocimiento de los distintos fenotipos y entidades clínicas existentes permitirán orientar mejor el estudio genético molecular necesario para precisar el diagnóstico etiológico.
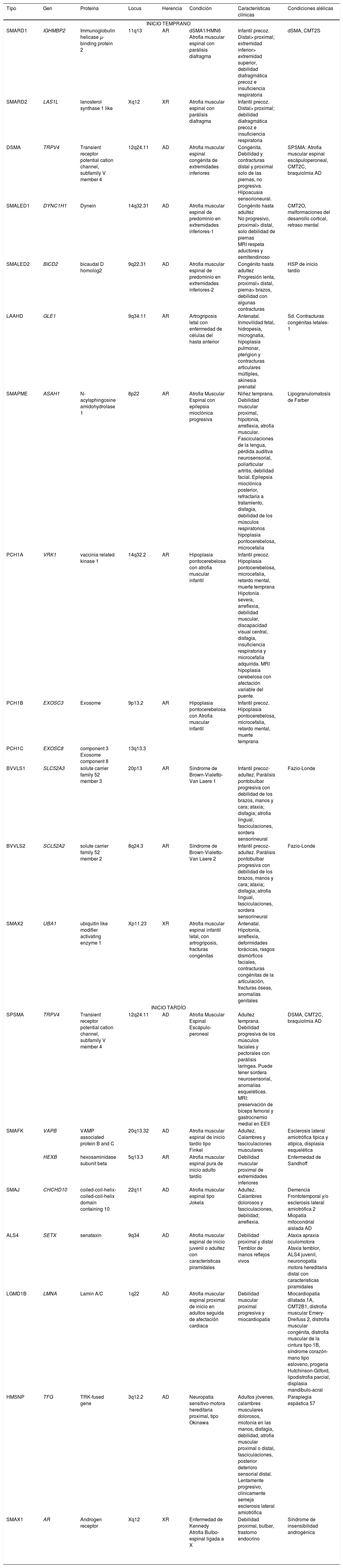
CLASIFICACIÓN DE LAS ATROFIAS MUSCULARES ESPINALES NO-5QLa característica definitoria de estas entidades es la presencia de una atrofia muscular espinal por compromiso de segunda motoneurona que se acompaña de diverso síntomas y distribución de debilidad que lo diferencia de la AME clásica. Existen a la fecha más de 18 genes involucrados en la etiología del AME no-5q, pero el listado aumenta constantemente, expandiendo incluso la presentación fenotípica de algunos genes ya conocidos (condiciones alélicas). La tabla 1 resume todas estas variables, destaca el patrón de herencia correspondiente, categoriza los diversos cuadros clínicos de acuerdo con la edad de inicio, distribución de la debilidad y enumera las condiciones alélicas conocidas. La figura 2 muestra los genes que comparten fenotipos y condiciones alélicas asociadas a compromiso de motoneuronas y sus sobreposiciones.
Resumen AME no 5q
| Tipo | Gen | Proteína | Locus | Herencia | Condición | Características clínicas | Condiciones alélicas |
|---|---|---|---|---|---|---|---|
| INICIO TEMPRANO | |||||||
| SMARD1 | IGHMBP2 | Immunoglobulin helicase μ-binding protein 2 | 11q13 | AR | dSMA1/HMN6 Atrofia muscular espinal con parálisis diafragma | Infantil precoz. Distal> proximal; extremidad inferior> extremidad superior, debilidad diafragmática precoz e insuficiencia respiratoria | dSMA, CMT2S |
| SMARD2 | LAS1L | lanosterol synthase 1 like | Xq12 | XR | Atrofia muscular espinal con parálisis diafragma | Infantil precoz. Distal> proximal; debilidad diafragmática precoz e insuficiencia respiratoria | |
| DSMA | TRPV4 | Transient receptor potential cation channel, subfamily V member 4 | 12q24.11 | AD | Atrofia muscular espinal congénita de extremidades inferiores | Congénita. Debilidad y contracturas distal y proximal solo de las piernas, no progresiva. Hipoacusia sensorioneural. | SPSMA: Atrofia muscular espinal escápuloperoneal, CMT2C, braquiolmia AD |
| SMALED1 | DYNC1H1 | Dynein | 14q32.31 | AD | Atrofia muscular espinal de predominio en extremidades inferiores-1 | Congénito hasta adultez No progresivo, proximal> distal, solo debilidad de piernas MRI respeta aductores y semitendinoso | CMT2O, malformaciones del desarrollo cortical, retraso mental |
| SMALED2 | BICD2 | bicaudal D homolog2 | 9q22.31 | AD | Atrofia muscular espinal de predominio en extremidades inferiores-2 | Congénito hasta adultez Progresión lenta, proximal> distal, pierna> brazos, debilidad con algunas contracturas | HSP de inicio tardío |
| LAAHD | GLE1 | 9q34.11 | AR | Artrogriposis letal con enfermedad de células del hasta anterior | Antenatal. Inmovilidad fetal, hidropesía, micrognatia, hipoplasia pulmonar, pterigion y contracturas articulares múltiples, akinesia prenatal | Sd. Contracturas congénitas letales-1 | |
| SMAPME | ASAH1 | N-acylsphingosine amidohydrolase 1 | 8p22 | AR | Atrofia Muscular Espinal con epilepsia mioclónica progresiva | Niñez temprana. Debilidad muscular proximal, hipotonía, arreflexia, atrofia muscular. Fasciculaciones de la lengua, pérdida auditiva neurosensorial, poliarticular artritis, debilidad facial. Epilepsia mioclónica posterior, refractaria a tratamiento, disfagia, debilidad de los músculos respiratorios hipoplasia pontocerebelosa, microcefalia | Lipogranulomatosis de Farber |
| PCH1A | VRK1 | vaccinia related kinase 1 | 14q32.2 | AR | Hipoplasia pontocerebelosa con atrofia muscular infantil | Infantil precoz. Hipoplasia pontocerebelosa, microcefalia, retardo mental, muerte temprana Hipotonía severa, arreflexia, debilidad muscular, discapacidad visual central, disfagia, insuficiencia respiratoria y microcefalia adquirida. MRI hipoplasia cerebelosa con afectación variable del puente. | |
| PCH1B | EXOSC3 | Exosome | 9p13.2 | AR | Hipoplasia pontocerebelosa con Atrofia muscular infantil | Infantil precoz. Hipoplasia pontocerebelosa, microcefalia, retardo mental, muerte temprana | |
| PCH1C | EXOSC8 | component 3 Exosome component 8 | 13q13.3 | ||||
| BVVLS1 | SLC52A3 | solute carrier family 52 member 3 | 20p13 | AR | Síndrome de Brown-Vialetto-Van Laere 1 | Infantil precoz-adultez. Parálisis pontobulbar progresiva con debilidad de los brazos, manos y cara; ataxia; disfagia; atrofia lingual, fasciculaciones, sordera sensorineural | Fazio-Londe |
| BVVLS2 | SCL52A2 | solute carrier family 52 member 2 | 8q24.3 | AR | Síndrome de Brown-Vialetto-Van Laere 2 | Infantil precoz-adultez. Parálisis pontobulbar progresiva con debilidad de los brazos, manos y cara; ataxia; disfagia; atrofia lingual, fasciculaciones, sordera sensorineural | Fazio-Londe |
| SMAX2 | UBA1 | ubiquitin like modifier activating enzyme 1 | Xp11.23 | XR | Atrofia muscular espinal infantil letal, con artrogriposis, fracturas congénitas | Antenatal. Hipotonía, arreflexia, deformidades torácicas, rasgos dismórficos faciales, contracturas congénitas de la articulación, fracturas óseas, anomalías genitales | |
| INICIO TARDÍO | |||||||
| SPSMA | TRPV4 | Transient receptor potential cation channel, subfamily V member 4 | 12q24.11 | AD | Atrofia Muscular Espinal Escápulo-peroneal | Adultez temprana. Debilidad progresiva de los músculos faciales y pectorales con parálisis laríngea. Puede tener sordera neurosensorial, anomalías esqueléticas. MRI: preservación de bíceps femoral y gastrocnemio medial en EEII | DSMA, CMT2C, braquiolmia AD |
| SMAFK | VAPB | VAMP associated protein B and C | 20q13.32 | AD | Atrofia muscular espinal de inicio tardío tipo Finkel | Adultez. Calambres y fasciculaciones musculares | Esclerosis lateral amiotrófica típica y atípica, displasia esquelética |
| HEXB | hexosaminidase subunit beta | 5q13.3 | AR | Atrofia muscular espinal pura de inicio adulto tardío | Debilidad muscular proximal de extremidades inferiores | Enfermedad de Sandhoff | |
| SMAJ | CHCHD10 | coiled-coil-helix-coiled-coil-helix domain containing 10 | 22q11 | AD | Atrofia muscular espinal tipo Jokela | Adultez. Calambres dolorosos y fasciculaciones, debilidad; arreflexia. | Demencia Frontotemporal y/o esclerosis lateral amiotrófica 2 Miopatía mitocondrial aislada AD |
| ALS4 | SETX | senataxin | 9q34 | AD | Atrofia muscular espinal de inicio juvenil o adultez con características piramidales | Debilidad proximal y distal Temblor de manos reflejos vivos | Ataxia apraxia oculomotora Ataxia temblor, ALS4 juvenil, neuronopatía motora hereditaria distal con características piramidales |
| LGMD1B | LMNA | Lamin A/C | 1q22 | AD | Atrofia muscular espinal proximal de inicio en adultos seguida de afectación cardíaca | Debilidad muscular proximal progresiva y miocardiopatía | Miocardiopatía dilatada 1A, CMT2B1, distrofia muscular Emery-Dreifuss 2, distrofia muscular congénita, distrofia muscular de la cintura tipo 1B, síndrome corazón-mano tipo esloveno, progeria Hutchinson-Gilford, lipodistrofia parcial, displasia mandíbulo-acral |
| HMSNP | TFG | TRK-fused gene | 3q12.2 | AD | Neuropatía sensitivo-motora hereditaria proximal, tipo Okinawa | Adultos jóvenes, calambres musculares dolorosos, miotonía en las manos, disfagia, debilidad, atrofia muscular proximal o distal, fasciculaciones, posterior deterioro sensorial distal. Lentamente progresivo, clínicamente semeja esclerosis lateral amiotrófica | Paraplegia espástica 57 |
| SMAX1 | AR | Androgen receptor | Xq12 | XR | Enfermedad de Kennedy Atrofia Bulbo-espinal ligada a X | Debilidad proximal, bulbar, trastorno endocrino | Síndrome de insensibilidad androgénica |
SMARD Spinal Muscular Atrophy with Respiratory Distress; DSMA Distal Spinal Muscular Atrophy; CMT Charcot Marie Tooth; SMALED Spinal Muscular Atrophy lower extremity Dominant; HSP Hereditary Spastic paraplegia; LAAHD lethal arthrogryposis with anterior horn cell disease; SMAPME Spinal Muscular Atrophy with Progressive Myoclonic Epilepsy; PCH Pontocerebellar Hypoplasia; BVVLS Brown-Vialetto-Van Laere Syndrome; SMAX X-linked Spinal Muscular Atrophy; SPSMA Scapuloperoneal Spinal Muscular Atrophy; SMAFK Spinal Muscular Atrophy Finkel Type; SMAJ Spinal Muscular Atrophy Jokela Type; ALS Amyotrophic lateral sclerosis; LGMD Limb Girdle Muscular Dystrophy; HMSNP Hereditary Motor and Sensory Neuropathy.

Espectro clínico de la ame no 5q
En la figura se destaca la heterogeneidad genética y la sobreposición fenotípica entre la AME, NSMH, HSP y ELA. AME: atrofia muscular espinal; NSMH: neuropatía sensitiva y motora hereditaria; ELA: esclerosis lateral amiotrófica; HSP: paraparesia espática hereditaria.
A continuación, mencionaremos algunas de estas patologías, teniendo en cuenta que no son necesariamente las más comunes, dado que es un grupo con una epidemiología no del todo precisada, pero que destacan entre otras. La AME con distrés respiratorio es una de las condiciones de inicio temprano de mayor severidad; las formas asociadas a TRPV4 hacen parte de uno de los espectros fenotípicos más amplio para un gen dentro del grupo; la neuronopatía por deficiencia del transportador de riboflavina destaca por tener tratamiento; la AME de predominio en extremidades inferiores tiene un patrón de compromiso muscular específico, con hallazgos característicos en la resonancia magnética muscular; la AME con hipoplasia ponto-cerebelosa presenta compromiso del neurodesarrollo de estructuras centrales y la atrofia muscular espinal y bulbar por ser una forma de inicio tardío ligado al cromosoma X con un marcado compromiso endocrinológico.
Atrofia Muscular Espinal con distres respiratorio (IGHMBP2)Las mutaciones en el gen que codifica para la proteína 2 de fijación de inmunoglobulina helicasa μ (IGHMBP2) son las responsables de la atrofia muscular espinal con dificultad respiratoria (SMARD1)17,18. Esta es una enfermedad autosómica recesiva caracterizada por debilidad muscular de predominio distal y dificultad respiratoria por parálisis diafragmática. Se instaura en el primer año de vida, con frecuencia entre las seis semanas y los seis meses de edad, progresando luego a una debilidad muscular generalizada que puede conducir a una tetraplejía completa y muerte temprana19,20. Estos pacientes a menudo nacen con bajo peso por restricción del crecimiento intrauterino y presentan deformidades, contracturas distales en manos y pies. Puede asociarse síntomas de compromiso del sistema nervioso autónomo y sensitivo con arritmias cardíacas, sudoración excesiva, retención urinaria, constipación, en ocasiones crisis epilépticas y paresia de nervios craneales. Se ha descrito variabilidad inter e intrafamiliar y formas de presentación tardías con neuropatías sin compromiso respiratorio así comouna forma axonal de Charcot-Marie-Tooth (CMT2S) que alcanza hasta el 12.9% de CMT recesivas21–23. Se ha determinado que el 1% de los pacientes con AME severo de inicio infantil se deben a mutación de IGHMBP220. Este gen codifica una helicasa de ADN/ARN implicada en la transcripción, el procesamiento de pre-ARNm, la traducción y la conmutación de clase de inmunoglobulina y su mutación conduce a la degeneración neuronal. Los mecanismos fisiopatológicos precisos para la enfermedad son desconocidos. Aunque se expresa en todos los tejidos, su disfunción parece afectar de forma selectiva y variable los nervios periféricos (afectación axonal), las motoneuronas inferiores y el sistema nervioso central. Para ayudar en el diagnóstico diferencial del SMARD1 dentro de las enfermedades de la motoneurona, Pitt et al24 propusieron una serie de criterios clínicos, histopatológicos y electrofisiológicos que en la actualidad conservan valor clínico, pero dados los avances en el diagnóstico genético, se debe replantear la necesidad de métodos invasivos como la biopsia de nervio (tabla 2). El futuro cercano de esta enfermedad parece promisorio ya que se han desarrollado nuevas tecnologías para el reemplazo eficaz del gen defectuoso por inyección intracerebroventricular de virus adeno asociados monocatenarios de serotipo 9 en el modelo animal de la enfermedad y los animales rescatados demostraron un aumento significativo de la masa muscular25,26.
Criterios diagnósticos SMARD1
| Criterios Clínicos | Criterios Histopatológicos | Criterios Electrofisiológicos |
|---|---|---|
| Bajo peso al nacer, debajo del 3er percentil | Reducción del diámetro de fibras mielinizadas en biopsia de nervio sural | Evidencia de denervación distal aguda o crónica |
| Inicio de los síntomas dentro de los primeros 3 meses de vida | Evidencia de degeneración de fibra mielinizadas en biopsias tomadas con 3-4 meses de separación | Evidencia de lentitud significativa (<70% del LIN) en uno o más nervios motores y/o sensitivos. |
| Debilidad diafragmática ya sea unilateral o bilateral | Sin evidencia de regeneración, que no sea la desmielinización, que pueda justificar la reducción en el tamaño de la fibra nerviosa | |
| Dependencia del ventilador en menos de un mes desde el inicio con incapacidad para destetar | ||
| Ausencia de dismorfología u otras afecciones |
Traducido de: Pitt M, Houlden H, Jacobs J, Mok Q, Harding B, Reilly M, et al. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain. 2003;126(12):2682–92.
Mutaciones en el gen TRPV4, que codifica un canal polimodal de calcio, son responsables de un sorprendente número de enfermedades con gran diversidad fenotípica27–29. Los tejidos y órganos afectados son generalmente el sistema esquelético y el sistema nervioso periférico30. Se han identificado mutaciones en TRPV4 en neuropatías sensitivo-motoras, AME congénita distal, AME escapuloperoneal y en Charcot- Marie-Tooth tipo 2C (CMT2C). Las mutaciones en este gen también son la causa de más de 5 displasias esqueléticas diferentes que tienen en común un tronco corto con platispondilia vertebral y escoliosis31. Algunos pocos pacientes presentan combinación de síntomas esqueléticos y neuropáticos. En ambos pueden existir una pérdida auditiva progresiva neurosensorial32. La mayoría de las personas diagnosticadas con un trastorno asociado a TRPV4 tienen un padre afectado. Sin embargo, dado que los fenotipos esqueléticos más severos pueden ser letales en la niñez (o en el útero), los niños con estos fenotipos probablemente tengan una variante patogénica de novo y padres no afectados33.
Particularmente, en la AME distal congénita, de herencia autosómica dominante, se incluyen trastornos que afectan principalmente a las extremidades en sus segmentos distales, se caracteriza por una atrofia muscular y debilidad que se limita principalmente a los miembros inferiores. Otros síntomas incluyen fasciculación de la lengua, contracturas de rodilla y cadera, escoliosis y pie bot. Algunos pacientes presentan ya al nacer una neuropatía motora distal grave y artrogriposis34. Es la única neuropatía dependiente de TRPV4 que no está asociada con síntomas sensoriales y no es progresiva35. En la AME escapuloperoneal existe una pérdida progresiva de las motoneuronas inferiores, asociada con debilidad muscular y atrofia proximal en la región de la cintura escapular (escápula alada) y músculos peroneos, además de disfunción laríngea (laringomalacia y anomalías de las cuerdas vocales con disfonía transitoria)36. La diversidad fenotípica de las mutaciones, a menudo ubicadas en los mismos dominios de la proteína, es uno de los rompecabezas más interesantes en la fisiopatología de este y otros canales iónicos.
Síndrome de Brown-Vialetto-Van-Laere o Neuronopatía por deficiencia del transportador de riboflavina (SLC52A2 y SLC52A3)Un trastorno notable en esta categoría es el Síndrome de Brown-Vialetto-Van Laere, se origina por mutaciones autosómicas recesivas en los genes transportadores de riboflavina o vitamina B2, SLC52A2 y SLC52A3. Se caracteriza por un fenotipo heterogéneo de aparición temprana, desde la primera a la tercera década, con parálisis pontobulbar progresiva, pérdida auditiva neurosensorial bilateral, debilidad muscular y amiotrofia debido a la degeneración de la motoneurona (MN). Aquellos con inicio temprano tienden a tener una progresión rápida de la enfermedad. En el síndrome de Fazio-Londe, causado por mutaciones en los mismos genes, la presentación clínica es la misma pero, sin la pérdida de auditiva y se considera que es un espectro de la misma enfermedad37.
La evidencia reciente muestra que el tratamiento con suplementos de riboflavina en alta dosis mejoran notablemente los síntomas clínicos, así como las anormalidades bioquímicas en los pacientes. Esto hace imprescindible estar alerta y tener conciencia de la forma de presentación y sospechar a tiempo este cuadro clínico, porque un tratamiento oportuno puede salvar la vida del paciente38–40.
Atrofia Muscular Espinal de predominio en extremidades inferiores (DYNC1H1 y BICD2)Producidas por mutaciones autosómicas dominantes de el gen DYNC1H1 y el gen BICD2 responsables de SMA-LED1 y SMA-LED2 respectivamente. Estas proteínas son importantes en el transporte axonal y las alteraciones funcionales identificadas revelan su papel en enfermedad de las motoneuronas. Se caracterizan por un inicio congénito o temprano, antes de los 5 años, con debilidad muscular predominantemente de los miembros inferiores, mayor en segmentos proximales y atrofia muscular sin compromiso sensitivo. Existe habitualmente un retraso de la marcha, marcha anadina, pérdida de reflejos distales. Los pacientes pueden presentar artrogriposis congénita, fasciculaciones, deformidades de los pies como pie calcáneo valgo, pie plano y pie valgo41,42. También se puede asociar a contracturas, displasia de cadera y/o hiperlordosis. El trastorno es estático o levemente progresivo. La resonancia magnética de los miembros superiores e inferiores demuestra un patrón característico de reemplazo de grasa e hipertrofia selectiva con afectación difusa de recto femoral, vasto lateral, vasto intermedio, tibial anterior, y gastrocnemio, con preservación relativa de los aductores medial semitendinoso y los músculos peroneos41.
Si bien las descripciones clínicas y patológicas iniciales destacaban el compromiso de células del asta anterior, el fenotipo en SMA-LED1 se ha ampliado para incluir la presencia de malformaciones corticales, discapacidad intelectual y espasticidad43. Hasta la fecha, no se ha establecido ninguna terapia de modificación de la enfermedad para SMA-LED.
Atrofia Muscular espinal con hipoplasia ponto-cerebelosa (VRK1, EXOSC3 y EXOSC8)Bajo el nombre de hipoplasias pontocerebelosa se conoce un grupo de diez trastornos neurodegenerativos severos, caracterizados por una degeneración espinocerebelosa, con malformación del vermis y lóbulo anterior del cerebelo, e hipoplasia severa del tronco encéfalo. El tipo 1 (PCH1), asociado a las mutaciones de los genes VRK1, EXOSC3 y EXOSC8, con patrón de herencia autosómico recesivo, es el que se asocia con enfermedad de la motoneurona. Las características clínicas asociadas con PCH1 son hipotonía severa, arreflexia, debilidad muscular, discapacidad visual central, disfagia, insuficiencia respiratoria y microcefalia adquirida con inicio clínico en los primeros meses de vida y muerte en la primera infancia. Algunas características adicionales son ataxia y nistagmo y si el inicio es prenatal, contracturas congénitas y polihidroamnios. La resonancia magnética cerebral muestra hipoplasia pontocerebelosa, incluido los hemisferios cerebelosos, juntos con afectación variable de la protuberancia y el cerebro. La electrofisiología muestra una neuropatía axonal motora sensitiva y denervación aguda en electromiografía44,45.
Atrofia Muscular espinal y bulbar (AR)La atrofia muscular espinal y bulbar, también conocida como enfermedad de Kennedy, es un desorden ligado al cromosoma X, causado por una expansión de repetición CAG en el gen del receptor de andrógenos (AR)46. La enfermedad se instaura entre los 18 y 64 años de edad, pero afecta principalmente hombres en la cuarta década de la vida, destacando dentro de la sintomatología el temblor, calambres, debilidad, atrofia muscular, disartria, disfagia y fasciculaciones periorales, también se puede asociar a signos y síntomas metabólicos y endocrinos como ginecomastia, impotencia y atrofia testicular47. Actualmente no existen tratamientos modificadores de la enfermedad, pero se encuentran en curso ensayos clínicos con terapias anti-androgénicas, con resultados promisorios48.
ORIENTACIÓN DIAGNÓSTICASe ha abordado la aproximación clínica que se debe realizar frente a un paciente en el que se sospecha alguna enfermedad relacionada con un compromiso primario de la segunda motoneurona, en particular, en el caso de las formas de inicio temprano sugerimos tener en cuenta el algoritmo propuesto en la figura 3. Indagar sobre qué otros sistemas u órganos están afectados, y reconocer las distintas entidades descritas en la tabla 1, permitirán un estudio genético molecular mejor orientado. Debido a la variabilidad clínica en el grupo de AME no 5q, la heterogeneidad fenotípica superpuesta y el locus genético, la secuenciación de nueva generación, en particular, los paneles de genes adaptados y la secuenciación completa del exoma/genoma son los métodos de diagnóstico más rentables que se utilizan en la actualidad49. El panel de genes evalúa en una sola prueba para todos los genes cuyas variantes alélicas patológicas se describen como causantes de AME no 5q, lo que aumenta la sensibilidad analítica a las pruebas de ADN y disminuye el tiempo para realizar el diagnóstico. El análisis de ligamiento y la secuenciación completa del exoma son las herramientas más ampliamente utilizadas para descubrir nuevos genes que causan enfermedades50.
CONCLUSIÓN
En la actualidad se dispone de métodos diagnósticos de nueva generación, con secuenciación masiva de genes y están al alcance de pacientes con diferentes realidades económicas; esto ha favorecido que muchos de los genes subyacentes a los trastornos de las motoneuronas hayan sido identificados, posibilitando un diagnóstico y pronóstico precoz. Si bien aun no existen tratamientos curativos para ninguna de estas patologías, es fundamental comprender que los primeros pasos para el desarrollo de intervenciones definitivas consisten en realizar diagnósticos de certeza y participar en los registros de pacientes para impulsar la investigación clínica. Adicionalmente, el avance en el conocimiento y la difusión del mismo, nos obliga a tener un alto nivel de sospecha en los casos en que tenemos pacientes que se salen del fenotipo de AME clásico, es decir, que presentan características clínicas adicionales descritas en esta revisión, particularmente en las formas de instauración temprana, dado que algunas de las patologías pueden tener intervenciones específicas, como en la neuronopatía por deficiencia del transportador de riboflavina, que modifican claramente el curso de la enfermedad.
Reconocer la historia natural de las diferentes formas de AME permite anticiparse a las complicaciones, no solo neurológicas sino también respiratorias, endocrinológicas, ortopédicas y funcionales entre otras, mejorando los estándares de cuidado y, por consiguiente, la calidad y expectativa de vida.
Declaración de interésEn relación con artículo Atrofia Muscular Espinal no ligada a cromosoma 5q, de la cual soy autor correspondiente, declaro no tener conflictos de interés.