






El isavuconazol es un nuevo azol, emparentado estructuralmente con el fluconazol y el voriconazol, que presenta una absorción por vía oral muy elevada, sin efecto de primer paso, y que no es interferida por la presencia de alimentos, modificaciones del pH gástrico, ni por la mucositis. Su distribución es muy elevada, probablemente también a líquido cefalorraquídeo, a pesar de que circula en plasma unido a proteínas en un porcentaje importante. Se elimina en su totalidad por el metabolismo mediante la isoenzima CYP3A4, de ahí que se recomiende evitar el uso conjunto de inductores potentes de esta isoenzima. Además, presenta capacidad de inhibir de forma moderada la CYP3A4 y de inducir de forma débil la CYP2B6 y la glucoproteína P. En cualquier caso, esta actividad inhibitoria parece inferior a la que presentan otros azoles, lo que deriva en un uso más sencillo en el manejo de las interacciones con otros fármacos, que es probablemente la ventaja más importante de este antifúngico.
Isavuconazole is a new azole, structurally related to fluconazole and voriconazole, that presents a very high oral absorption with no first-pass effect which is not interfered by the presence of food, gastric pH modifications, or mucositis. Its distribution volume is very high, probably also to cerebrospinal fluid, in spite of the fact that it circulates highly bound to plasma proteins. It is extensively metabolized through the CYP3A4 isoenzyme. Due to this reason, it is recommended to avoid co-administration with strong CYP3A4 inducers. In addition, isavuconazole may inhibit CYP3A4. Moreover, it may induce CYP2B6 and P-glycoprotein. Interestingly, this inhibitory activity seems to be lower compared to other azoles. Therefore, the management of any interaction with other medicines is easier, which is probably the most important advantage of this antifungal.
Las enfermedades infecciosas sistémicas producidas por hongos continúan siendo todo un problema debido a su mortalidad, a pesar de que el arsenal terapéutico disponible ha aumentado de forma considerable en los últimos años. Por ello, resulta necesario dar la bienvenida a otro nuevo fármaco, el isavuconazol, que por sus propiedades puede estar llamado a ocupar un lugar relevante en el tratamiento de estas infecciones, tal y como se describe en los diferentes apartados de esta monografía. En este artículo se detallarán de forma aplicada los diferentes aspectos de su farmacología.
El isavuconazol presenta baja solubilidad en agua, por lo que con la finalidad de facilitar su administración evitando el uso de ciclodextrina, común en las restantes formulaciones intravenosas de otros azoles, se desarrolló como isavuconazonio (BAL8557), que incorpora al isavuconazol un grupo amoniocarboxilo formando una sal de triazolio que aumenta su solubilidad. Tras la administración se produce la hidrólisis en el plasma, en la que participa, como enzima más importante, la butilcolinesterasa (fig. 1). Por tanto, el isavuconazol es la fracción activa del profármaco isavuconazonio (BAL8557). Su nombre químico es 1-[(2R,3R)-3-[4-(4-cianofenil)-1,3-tiazol-2-il]-2-(2,5-difluorofenil)-2-hidroxibutil]-4-[1-({metil[3-({[2-(metilamino)acetil]oxi}metil)piridin-2-il]carbamoil}oxi)etil]-1H-1,2,4-triazol-4-ium monosulfato. Esta estructura química presenta similitudes con la estructura del fluconazol y del voriconazol7,12,13.
Mecanismo de acción y actividad antifúngica y del isavuconazol (BAL4815).")
Al igual que otros azoles, el isavuconazol produce la inhibición de algunos de los pasos que conducen a la síntesis del ergosterol, componente estructural fundamental de la membrana del hongo. Entre ellos, la reacción en la que interviene la enzima 14-lanosterol-desmetilasa (CYP51), codificada por el gen ERG11, que es dependiente del CYP450 del hongo, y que retira el grupo metilo en posición 14 del lanosterol. La inhibición de esta enzima produce una alteración de la estructura de la membrana y la pérdida de sus funciones. Además, se produce la acumulación de los precursores que termina produciendo toxicidad directa sobre las células fúngicas. El isavuconazol muestra una afinidad elevada por la mitad hem de la enzima 14-alfa-desmetilasa a través del nitrógeno presente en posición 46,11. El tamaño de la molécula facilita su orientación correcta y, probablemente, al igual que el voriconazol1, la fijación al centro catalítico hidrofóbico de la enzima, lo que contribuiría a proporcionar un espectro antifúngico amplio. Se han descrito valores de IC50 de 0,043mg/l frente a la 14 alfa-desmetilasa extraída de Candida albicans, mientras que estos valores se sitúan en 4mg/l cuando la enzima es de origen murino, de ahí que se pueda señalar la selectividad de la inhibición y la posibilidad de que la capacidad inhibitoria del CYP450 humano sea solo moderada2.
FarmacocinéticaEl isavuconazol, administrado en forma de isavuconazonio, es rápidamente convertido (vida media inferior a 1,5min) en la fracción activa isavuconazol, por lo que el isavuconazonio es prácticamente indetectable después de la administración por vía oral.
Tras la administración sin dosis de carga, el estado de equilibrio se alcanza aproximadamente 14 días después de la administración del fármaco, lo que es consistente con la semivida de eliminación que oscila entre 100 y 130h. La administración de una dosis de carga de 200mg tres veces al día los dos primeros días de tratamiento permite alcanzar el estado de equilibrio en el tercer día.
La variabilidad interindividuo en los valores de la Cmax (concentración plasmática máxima) y AUC (área bajo la curva) está en el 20-30% con independencia de la vía de administración. En estudios de farmacocinética poblacional la variabilidad entre sujetos para el AUC fue del 58% y la intersujetos del 45%. No existieron diferencias entre la farmacocinética evaluada en sujetos sanos y en pacientes. En general, con la dosis recomendada, los valores de Cmin (concentración plasmática mínima) de isavuconazol alcanzados se sitúan alrededor de 3μg/ml, cifra que se encuentra por encima de los valores de concentración mínima inhibitoria (CMI) críticos de 1-2μg/ml2.
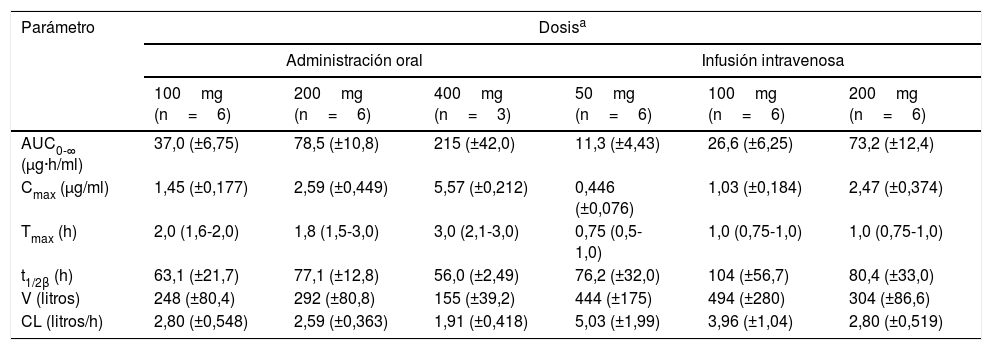
El comportamiento farmacocinético del isavuconazol ha sido evaluado en una cohorte de voluntarios sanos que recibieron una dosis única en régimen ascendente de 100, 200, 400mg por vía oral, y 50, 100 y 200mg por vía intravenosa, obteniéndose las curvas de niveles plasmáticos y los parámetros farmacocinéticos que se incluyen en la tabla 116.
Parámetros farmacocinéticos del isavuconazol tras la administración de una dosis única, oral e intravenosa, de isavuconazonio
| Parámetro | Dosisa | |||||
|---|---|---|---|---|---|---|
| Administración oral | Infusión intravenosa | |||||
| 100mg (n=6) | 200mg (n=6) | 400mg (n=3) | 50mg (n=6) | 100mg (n=6) | 200mg (n=6) | |
| AUC0-∞ (μg·h/ml) | 37,0 (±6,75) | 78,5 (±10,8) | 215 (±42,0) | 11,3 (±4,43) | 26,6 (±6,25) | 73,2 (±12,4) |
| Cmax (μg/ml) | 1,45 (±0,177) | 2,59 (±0,449) | 5,57 (±0,212) | 0,446 (±0,076) | 1,03 (±0,184) | 2,47 (±0,374) |
| Tmax (h) | 2,0 (1,6-2,0) | 1,8 (1,5-3,0) | 3,0 (2,1-3,0) | 0,75 (0,5-1,0) | 1,0 (0,75-1,0) | 1,0 (0,75-1,0) |
| t1/2β (h) | 63,1 (±21,7) | 77,1 (±12,8) | 56,0 (±2,49) | 76,2 (±32,0) | 104 (±56,7) | 80,4 (±33,0) |
| V (litros) | 248 (±80,4) | 292 (±80,8) | 155 (±39,2) | 444 (±175) | 494 (±280) | 304 (±86,6) |
| CL (litros/h) | 2,80 (±0,548) | 2,59 (±0,363) | 1,91 (±0,418) | 5,03 (±1,99) | 3,96 (±1,04) | 2,80 (±0,519) |
AUC: área bajo la curva; Cmax: concentración plasmática máxima; Tmax: tiempo de concentración plasmática máxima; t1/2 b: semivida de eliminación; V: volumen de distribución; CL: aclaramiento.
Los valores son presentados como media y desviación estándar, exceptuando la Tmax, que se presenta como mediana y límites.
La Cmax tras la administración por vía oral se alcanzó entre 2 y 3h después de la administración. Los valores de Cmax y de AUC no fueron enteramente proporcionales a la dosis administrada ya que resultaron ligeramente superiores con los aumentos de dosis. La semivida de eliminación fue muy prolongada, 56-70h, en la vía oral y 76-104h en la intravenosa. El volumen de distribución resultó muy elevado, 155-292l y 304-494l en la vía oral e intravenosa, respectivamente. El aclaramiento sistémico fue de 1,9-2,8l/h en la vía oral y de 2,8-5l/h en la intravenosa. Se eliminó por la orina en forma no modificada el 0,4% de la dosis administrada. La biodisponibilidad después de la administración oral prácticamente completa (98%).
También se ha evaluado el comportamiento farmacocinético del isavuconazol después de la administración de una dosis múltiple por vías oral e intravenosa en voluntarios sanos. Se administró una dosis de carga de 100 y 200mg seguida por dosis de mantenimiento diaria de 50 y 100mg, respectivamente. Esta pauta se utilizó durante 21 días en el caso de la vía oral y 15 días en el de la intravenosa. Existió proporcionalidad en los valores de Cmax y AUC en relación con la dosis administrada. El AUC se elevó a lo largo de los días de tratamiento entre 4 y 5 veces respecto a la inicial, estimándose una semivida de eliminación de 84,5-117h tras la última administración. El aclaramiento sistémico fue reducido: 2,4-4,1l/h y el volumen de distribución resultó elevado (308-542l)17.
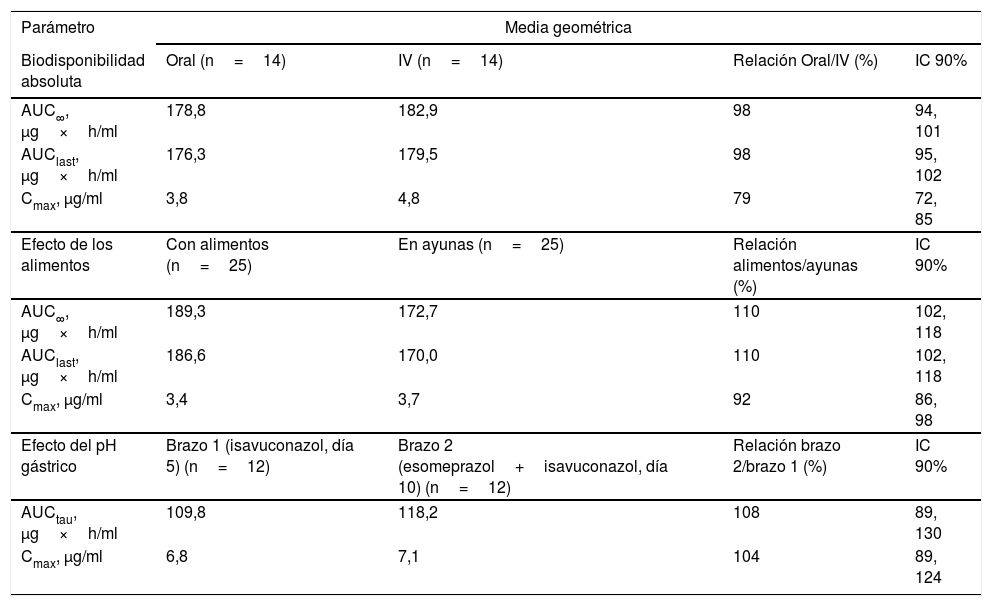
Se han realizado dos ensayos clínicos de dosis única, cruzados, aleatorizados, y un ensayo abierto de dosis múltiple en voluntarios sanos, para determinar la biodisponibilidad absoluta, el efecto de la ingesta de alimentos y el de los cambios en el pH gástrico, producidos por la administración de 40mg/día de esomeprazol. Se administraron 400mg de isavuconazol por vías oral e intravenosa para el estudio de la biodisponibilidad y 200mg tres veces al día durante dos días, seguido por 200mg/día durante tres días, en los estudios de los alimentos y cambios del pH gástrico. Tal y como se muestra en la tabla 2, la biodisponibilidad absoluta fue del 98% (IC: 94, 101) para el AUC0-∞, y del 78% (IC 90%: 72, 85) para la Cmax. La relación AUC alimento/ayunas fue del 110% (IC 90%: 102, 118) y 92% (IC 90%: 86, 98) para el AUC y Cmax, respectivamente, aunque el valor de la tmax se retrasó a 5h desde las 3h, observado en la administración en ayunas. No se produjeron modificaciones de la biodisponibilidad por efecto del esomeprazol, y la relación AUC y Cmax con y sin esomeprazol fue del 108% (IC 90%: 89, 130) y 105% (IC 90%: 89, 124), respectivamente15 (tabla 2).
Farmacocinética plasmática del isavuconazol tras la administración en diferentes condiciones
| Parámetro | Media geométrica | |||
|---|---|---|---|---|
| Biodisponibilidad absoluta | Oral (n=14) | IV (n=14) | Relación Oral/IV (%) | IC 90% |
| AUC∞, μg×h/ml | 178,8 | 182,9 | 98 | 94, 101 |
| AUClast, μg×h/ml | 176,3 | 179,5 | 98 | 95, 102 |
| Cmax, μg/ml | 3,8 | 4,8 | 79 | 72, 85 |
| Efecto de los alimentos | Con alimentos (n=25) | En ayunas (n=25) | Relación alimentos/ayunas (%) | IC 90% |
| AUC∞, μg×h/ml | 189,3 | 172,7 | 110 | 102, 118 |
| AUClast, μg×h/ml | 186,6 | 170,0 | 110 | 102, 118 |
| Cmax, μg/ml | 3,4 | 3,7 | 92 | 86, 98 |
| Efecto del pH gástrico | Brazo 1 (isavuconazol, día 5) (n=12) | Brazo 2 (esomeprazol+isavuconazol, día 10) (n=12) | Relación brazo 2/brazo 1 (%) | IC 90% |
| AUCtau, μg×h/ml | 109,8 | 118,2 | 108 | 89, 130 |
| Cmax, μg/ml | 6,8 | 7,1 | 104 | 89, 124 |
AUC∞: área bajo la curva de concentración plasmática/tiempo, extrapolada al infinito; AUClast: área bajo la curva de concentración plasmática/tiempo, entre cero y última concentración cuantificable; AUCtau: área bajo la curva de concentración plasmática/tiempo, durante el intervalo posológico; Cmax: concentración plasmática máxima; IC: intervalo de confianza; IV: intravenosa.
Tal y como se ha señalado, el volumen de distribución del isavuconazol es muy elevado, al igual que la fijación a proteínas plasmáticas, principalmente albúmina (99,3%). Estos valores no se modifican en pacientes con insuficiencia renal. Considerando el alto volumen de distribución del fármaco, que se sitúa en unos 450l, las interacciones entre fármacos debidas a desplazamiento por proteínas no son previsibles2.
En modelos experimentales el isavuconazol alcanza concentraciones elevadas y mantenidas en comparación con las concentraciones plasmáticas en algunos tejidos como el riñón, a pesar de que prácticamente no se elimina por vía renal21.
No existe información sobre la penetración en líquido cefalorraquídeo, aunque el isavuconazol se ha mostrado activo en el tratamiento de la criptococosis meníngea del ratón y en el de algunos pacientes con infecciones localizadas del sistema nervioso central18, lo que indica que el fármaco es capaz de atravesar la barrera hematoencefálica.
MetabolismoLas principales vías del metabolismo del isavuconazol son la oxidación con hidrólisis del grupo ciano, la oxidación de la forma carbomoil, y la rotura del anillo triazol. Tras esta se produce un metabolismo adicional principalmente por oxidación y subsecuente glucuronidación y conjugación con N-acetilcisteína. Ninguno de los metabolitos presenta valores de AUC superiores al 10% del fármaco original2.
Los estudios in vitro/in vivo indican que tanto CYP3A4 como CYP3A5 y la uridin-difosfato glucuronosiltransferasa (UGT) están involucradas en el metabolismo del isavuconazol.
EliminaciónLa semivida de eliminación plasmática del isavuconazol es de 100-130h. El aclaramiento total tras una dosis única administrada por vía intravenosa u oral se sitúa en aproximadamente 2l/h. El aclaramiento renal oscila entre 7,4 y 14ml/h.
Tras la administración oral de sulfato de isavuconazonio marcado a sujetos sanos, se recuperó una media del 46,1% de la dosis radioactiva en las heces y el 45,5% en la orina. La excreción renal de isavuconazol intacto fue inferior al 1% de la dosis administrada. El producto de degradación inactivo se elimina principalmente por el metabolismo y la posterior excreción renal de los metabolitos2.
Se ha descrito un modelo de farmacocinética poblacional, realizado con los resultados obtenidos en un ensayo clínico en fase 3 que fue realizado para evaluar la eficacia y tolerabilidad del isavuconazol en el tratamiento de pacientes con insuficiencia renal que presentaban infecciones invasivas producidas por diversos hongos filamentos, hongos dimórficos y levaduras. Las covariables evaluadas fueron las siguientes: índice de masa corporal, peso, raza, filtrado glomerular y aclaramiento. Entre los resultados más destacables se señaló que mientras el peso era una covariable que afectaba al aclaramiento, las modificaciones del filtrado glomerular no tenían influencia. Los parámetros farmacocinéticos y la variabilidad del aclaramiento intersujeto fueron similares y no se relacionaron con el tipo de infección. El análisis PK/PD mostró que el régimen posológico utilizado era suficiente para alcanzar valores de AUC/CMI situados en 50,5 para Aspergillus (CLSI CMI de 0,5mg/l), 270 y 5053 para C. albicans (CMI: 0,125 y 0,004mg/l, respectivamente) y 312 para otras especies de Candida (CMI: 0,125mg/l)9.
Farmacocinética en situaciones especialesInsuficiencia renalSe han comparado los parámetros farmacocinéticos obtenidos tras la administración de 200mg de isavuconazol a sujetos sanos y a pacientes que presentaban diferentes grados de insuficiencia renal. Además, los pacientes sometidos a hemodiálisis recibieron dos dosis de 200mg, una antes (día 1) y otra inmediatamente después de finalizado el procedimiento (día 15). Los valores de Cmax resultaron un 4% superiores en pacientes con insuficiencia renal leve y 7, 14 y 21% inferiores en los pacientes que presentaban insuficiencia renal moderada, grave y terminal, respectivamente. Los pacientes en diálisis presentaban valores de AUC0-72 un 30% superiores al finalizar esta técnica. Las AUC (AUC∞ y AUClast) no fueron diferentes en los sujetos control; no existieron diferencias con significación estadística en la semivida de eliminación, aclaramiento o volumen de distribución, por lo que los autores concluyeron que no se requiere ajuste de la dosis en relación con la función renal del paciente, ni cuando se realizan técnicas de diálisis20.
Insuficiencia hepáticaTras haber administrado una dosis única de 100mg de isavuconazol a 32 pacientes con insuficiencia hepática leve (Child-Pugh A) y a un número similar de pacientes con insuficiencia hepática moderada (Child-Pugh B) (16 pacientes por vía intravenosa y 16 pacientes por vía oral por cada grupo), la exposición sistémica media (AUC) aumentó un 64% en el grupo de Child-Pugh A, y un 84% en el grupo de Child-Pugh B, con relación a 32 sujetos sanos emparejados por edad y peso. Los valores Cmax eran un 2% y un 30% inferiores en los pacientes del grupo de Child-Pugh A y B, respectivamente. La evaluación farmacocinética poblacional del isavuconazol en sujetos sanos y en pacientes con disfunción hepática leve o moderada mostró que la población con insuficiencia hepática leve y moderada presentaba valores de aclaramiento del isavuconazol un 40 y 48% menores, respectivamente, en comparación con la población sana. Según estos resultados no se considera necesario el ajuste de la dosis en pacientes con insuficiencia hepática de leve a moderada.
El isavuconazol no se ha estudiado en pacientes con insuficiencia hepática grave (Child-Pugh clase C). No se recomienda el uso en estos pacientes a menos que el posible beneficio supere el riesgo3,4.
Otras situacionesSe ha evaluado la farmacocinética del isavuconazol en 56 pacientes con mucositis incluidos en los ensayos clínicos, resultando la biodisponibilidad similar a la observada entre los pacientes sin mucositis. Además, la mortalidad y la respuesta clínica global resultaron similares a la de los pacientes sin mucositis10.
InteraccionesEl isavuconazol es un sustrato e inhibidor moderado de la CYP3A4; además puede inducir la isoenzima CYP2B6. La capacidad del isavuconazol de inhibir la glucoproteína BCRP, OCT2 y UGT es ligera3.
CYP3A4La coadministración de 100mg/día de isavuconazol junto con 600mg de rifampicina, inductor de la actividad de la isoenzima CYP3A4, redujo el AUC y la Cmax del isavuconazol el 90 y 75%, respectivamente. Al contrario, el ketoconazol, potente inhibidor de la CYP3A4, administrado a dosis de 200mg/12h, aumentó el AUC y la Cmax del isavuconazol un 422 y 9%, respectivamente19.
La administración de isavuconazol junto con midazolam (3mg) o etinil estradiol/noretindrona (35μg/1mg), sustratos de la CYP3A4, aumentó un 103% el AUC de midazolam y 8/16% el de etinil estradiol/noretindrona. Además, la Cmax aumentó un 72, 14, y 6%, respectivamente.
Estos resultados confirman que el isavuconazol es un sustrato e inhibidor moderado de CYP3A4, lo que implica que puede causar un aumento de las concentraciones en plasma de los medicamentos que son sustratos de la CYP3A4/5.
Por otra parte, la administración conjunta de isavuconazol con inductores potentes de la CYP3A4/5, como rifampicina, rifabutina, carbamazepina, barbitúricos de acción prolongada (como el fenobarbital), fenitoína y hierba de San Juan o con inductores moderados de la CYP3A4/5 como efavirenz, nafcilina y etravirina está contraindicada, dado que estos medicamentos pueden disminuir significativamente las concentraciones en plasma del isavuconazol, reduciendo su eficacia.
La administración junto con inductores leves de la CYP3A4/5 como aprepitant, prednisona y pioglitazona puede causar una disminución entre leve y moderada de los niveles en plasma del isavuconazol, por lo que se debe evitar la administración conjunta a menos que el posible beneficio supere el riesgo.
La administración junto con dosis elevadas de ritonavir (200mg o más dos veces al día) está contraindicada, dado que el ritonavir a dosis altas puede inducir la CYP3A4/5 y disminuir las concentraciones en plasma del isavuconazol.
La administración conjunta de isavuconazol con inhibidores muy potentes de la CYP3A4/5, como el ketoconazol, está contraindicada, puesto que este medicamento puede aumentar significativamente las concentraciones en plasma del isavuconazol.
Para los inhibidores potentes de la CYP3A4, como lopinavir/ritonavir, se observó el doble de aumento en la exposición al isavuconazol. Para otros inhibidores potentes de la CYP3A4, como la claritromicina, indinavir y saquinavir, cabe esperar un efecto menos pronunciado, basado en su potencia relativa. No es necesario ajustar la dosis del isavuconazol al administrarlo junto con inhibidores potentes de la CYP3A4/5; no obstante, se debe proceder con precaución dado que las reacciones adversas a los medicamentos pueden aumentar3.
InmunosupresoresSe han realizado algunos estudios para evaluar el efecto del isavuconazol sobre la farmacocinética de algunos inmunosupresores de uso habitual, como la ciclosporina, el ácido micofenólico, la prednisolona, el sirolimus y el tacrolimus en voluntarios sanos8. Los pacientes recibieron una dosis única por vía oral de los siguientes medicamentos: ciclosporina (300mg; n=24), micofenolato mofetilo (1.000mg; n=24), prednisolona (20mg; n=21), sirolimus (2mg; n=22), y tacrolimus (5mg; n=24), con y sin la administración de isavuconazol oral (200mg 3 veces diarias, dos días y 200mg una vez al día posteriormente). La coadministración generó un aumento de AUC0-∞ de tacrolimus, sirolimus, y ciclosporina del 125, 84, y 29%, respectivamente. En el caso del ácido micofenólico y la prednisolona el aumento fue del 35 y 8%, respectivamente. La Cmax de tacrolimus, sirolimus, y ciclosporina aumentó el 42, 65, y 6%, respectivamente, mientras que la del ácido micofenólico y la prednisolona fue un 11 y 4% inferior, respectivamente. La farmacocinética del isavuconazol no fue modificada por ninguno de los fármacos. Los autores concluyen que la intensidad del efecto inhibitorio del isavuconazol sobre la isoenzima CYP3A4 parece inferior a la de otros azoles8.
Se ha evaluado de forma específica la influencia del isavuconazol sobre la farmacocinética del tacrolimus en 55 receptores de un trasplante de órgano sólido. Se realizó un análisis retrospectivo de pacientes que habían iniciado profilaxis antifúngica con isavuconazol con una duración igual o superior a 21 días y que también habían recibido como profilaxis frente al rechazo tacrolimus y que, además, disponían de seguimiento clínico durante al menos 40 días desde la suspensión del antifúngico. Se calculó la relación concentración de tacrolimus/dosis (C/D) normalizada por el peso corporal, de acuerdo con la siguiente fórmula: [tacrolimus Concentración mínima/(dosis diaria total/peso corporal actual)]. Se realizó una comparación de estos datos obtenidos entre los días 8-21 de tratamiento con isavuconazol y los días 29-60 cuando el antifúngico ya había sido suspendido. La mediana de la relación C/D de tacrolimus fue más elevada durante el tratamiento con isavuconazol (125 y 120ng/ml por mg/kg, respectivamente; p=0,037). La relación C/D de tacrolimus se redujo una media del 13% después de la suspensión del antifúngico. La dosis media de tacrolimus fue aumentada 1,3 veces tras la suspensión del isavuconazol. Los autores concluyeron que la dosis de tacrolimus no debe ser ajustada de forma sistemática cuando se inicia la administración de isavuconazol, sino una vez realizados los controles de las concentraciones plasmáticas del fármaco, circunstancia que lo diferencia de lo que sucede con otros antifúngicos azólicos14.
La comparación del impacto de los diferentes azoles, incluido el isavuconazol, en la Cmin o el AUC de la ciclosporina, el tacrolimus o el sirolimus señala que el manejo de los tres inmunosupresores con isavuconazol es bastante más sencillo que con los restantes azoles, que producen un aumento de la Cmin y/o del AUC mucho más importante8 (tabla 3).
Interacciones entre antifúngicos triazoles y algunos inmunosupresores: ciclosporina A, sirolimus y tacrolimus
| CYP3A4 Sustrato | Isavuconazol | Fluconazol | Itraconazol | Posaconazol | Voriconazol |
|---|---|---|---|---|---|
| Ciclosporina | ↑ 1,3a | ↑ 2-3a | ↑ 2,7b | ↑b,c | ↑ 1,7a |
| Sirolimus | ↑ 1,8a | ↑ 3-4,7b | NA | ↑ 7,9a | ↑ 11a |
| Tacrolimus | ↑ 2,3a | NA | ↑ 5,6b | ↑ 3,6a | ↑ 3a |
NA: no aplicable.
Tal y como se ha señalado, el isavuconazol no es sustrato de ninguna otra isoenzima diferente al CYP3A4, a diferencia del voriconazol y el fluconazol que sí lo son del CYP9 y CYP19, de donde se derivan el mayor número de interacciones, además de la posibilidad de que sufran alteraciones en la eliminación en relación con polimorfismos genéticos del paciente. En este sentido se ha descartado el riesgo de interacción entre el isavuconazol y la warfarina, sustrato del CYP2C95, en un estudio de dosis múltiples de isavuconazol (200mg/día), más dosis de carga convencional, con y sin la administración de 20mg de warfarina sódica.
De idéntico modo se ha descartado riesgo de interacciones con una dosis única de repaglinida (CYP2C8/CYP3A4; 0,5mg; n=24), cafeína (CYP1A2; 200mg; n=24), dextrometorfano (CYP2D6/CYP3A4; 30mg; n=24), y metadona (CYP2B6/CYP2C19/CYP3A4; 10mg; n=23)22.
Estos resultados señalan la ausencia de efecto del isavuconazol sobre los fármacos que se metabolizan a través de las isoenzimas CYP1A2, CYP2C8, o CYP2D6, mientras que se comporta como un inductor ligero de la CYP2B6, por lo que reduce la biodisponibilidad de bupropión (CYP2B6; 100mg; n=24). La exposición sistémica de los medicamentos metabolizados por la CYP2B6 (ciclofosfamida, efavirenz) puede disminuir al administrarse junto con el isavuconazol.
Proteínas transportadorasSe ha evaluado el impacto del isavuconazol sobre algunos sustratos de las proteínas transportadoras más importantes: proteína de resistencia del cáncer de mama (BCRP), proteína de eflujo de múltiples fármacos y tóxicos-1 (MATE1), transportador de aniones orgánicos 1 y 3 (OAT1/OAT3), polipéptido transportador de aniones orgánicos 1B1 (OATP1B1), transportador de cationes orgánicos (OCT1 y OCT2), y glucoproteína P (P-gp) en sujetos sanos. Estos recibieron dosis única de atorvastatina (20mg; sustrato de OATP1B1 y P-gp), digoxina (0,5mg; sustrato P-gp), metformina (850mg; sustrato OCT1, OCT2, y MATE1), o metotrexato (7,5mg; sustrato de BCRP, OAT1, y OAT3) con o sin isavuconazol (200mg 3 veces al día, durante 2 días y 200mg una vez al día después). La coadministración del isavuconazol aumentó el AUC de la atorvastatina, la digoxina y la metformina un 137% (129, 145), 125% (117, 134) y 152% (138, 168), respectivamente, mientras que las Cmax aumentaron un 103% (88, 121), 133% (119, 149), y 123% (109, 140), respectivamente. Los parámetros de metotrexato no se modificaron. Los hallazgos demuestran que el isavuconazol es un débil inhibidor de la P-gp, y probablemente de OCT1, OCT2, MATE1, pero no de BCRP, OATP1B1, OAT1, u OAT323.
Conflicto de interesesJ.R. Azanza ha participado en diversos proyectos como consultor e impartido conferencias y ponencias con MSD, Pfizer y Astellas. El resto de los autores declaran no tener ningún conflicto de intereses.

