













Introducción
El proceso de detoxificación de los fármacos (conversión en hidrosolubles) se produce en el hígado, lo que determina que sea el principal órgano diana de posibles reacciones adversas. Aunque es considerable el número de fármacos que se han relacionado con hepatotoxicidad, esta complicación no constituye un problema clínico habitual a causa de la baja incidencia de reacciones idiosincrásicas (entre 1/1.000 y 1/100.000 prescripciones). No obstante, el 10-15% de las hepatitis fulminantes son debidas a fármacos, así como un 2-5% de los síndromes ictéricos que ingresan en el hospital. La frecuencia con la que ciertos medicamentos causan un determinado tipo de hepatopatía se recaba de los informes de las admisiones hospitalarias y de los grupos diagnósticos, observándose que abundan las discrepancias entre los distintos estudios1-3.
La reacción hepatotóxica puede remedar cualquier síndrome agudo o crónico y constituye un problema habitual de diagnóstico diferencial. Debido a la ausencia de un marcador específico de hepatopatía tóxica, la atribución etiológica a un medicamento comporta un proceso de exclusión de otras causas lesivas. La hepatotoxicidad es uno de los motivos más frecuentes de retirada de fármacos del mercado; a pesar de que los estudios para detectar posibles efectos tóxicos en las fases preclínica y clínica (antes de la comercialización de un nuevo compuesto) hayan sido tan exhaustivos, no pueden ser representativos de la ingente potencial heterogeneidad de pacientes tributarios del tratamiento y, por ello, las reacciones idiosincrásicas con frecuencia se detectan tan sólo después de que una amplia población haya sido expuesta al fármaco1-4.
Metabolismo hepático de los fármacos
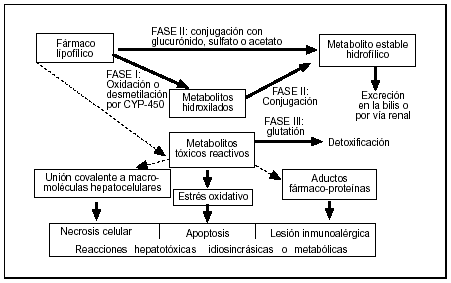
La acción fisiológica o tóxica de los distintos compuestos xenobióticos lipofílicos perdura en función del tiempo de permanencia en el organismo, el cual depende de las enzimas hepáticas que catalizan su conversión en derivados hidrofílicos. El principal sistema enzimático es el citocromo P450 (CYP) por su capacidad para actuar como una oxidasa incorporando oxígeno a los sustratos lipofílicos (oxidación), presentando una especificidad de sustrato que reside en su región apoproteica. La metabolización de algunos medicamentos precisa de varios pasos, cada uno mediado por un CYP diferente, hasta convertirse en una molécula terapéuticamente inactiva. Sin embargo, el metabolismo de numerosos fármacos origina intermediarios biológicamente activos que son responsables de las acciones farmacológicas o tóxicas; estas reacciones se denominan de fase I o «toxificadoras», ya que liberan radicales químicamente activos. Cuando dichos metabolitos no son inactivados mediante reacciones de fase II (ya sea por conjugación con ácido glucurónico o sulfúrico, por acetilación o acoplándose al glutatión), tienen la capacidad de dañar directamente a macromoléculas (proteínas o ácidos nucleicos) celulares1,5. Por tanto, la eliminación de un medicamento depende de una familia de enzimas y, cuando éstas son deficientes o están saturadas, se derivan consecuencias tóxicas (fig. 1).

Figura 1. Biotransformación hepática de los fármacos en compuestos polares y mecanismos de hepatotoxicidad (líneas gruesas representan vías metabólicas principales). Adoptada de Lee, 1995.
Los metabolitos originados en la fase I pueden actuar a su vez como haptenos y dar lugar a una reacción inmunoalérgica expresada como una respuesta humoral (anticuerpos), celular (linfocitos T) o mixta. La reacción inmune puede ir dirigida contra el fármaco, la proteína transportadora (determinantes autoantigénicos) o neoantígenos originados por la combinación de ambos. Por último, el metabolito reactivo puede inducir un trastorno de la inmunorregulación causante de una hepatitis autoinmune típica, con positividad sérica para autoanticuerpos no específicos de órgano (como en la hepatitis por halotano) o específicos de órgano como los anti-LKM que reaccionan contra distintas enzimas microsomales1,5.
Determinados factores relacionados con el huésped (predisposición genética, edad, inducción enzimática o enfermedades asociadas) favorecen un desequilibrio entre la generación de metabolitos tóxicos en la fase I y la tasa de su posterior inactivación. No siempre es posible identificar las causas específicas que alteran este equilibrio, por lo que las reacciones hepatotóxicas por ahora son inevitables.
Definición de la lesión hepática y consecuencias de la hepatotoxicidad
Se ha establecido en reuniones de consenso que debe producirse al menos una de las siguientes alteraciones de la bioquímica hepática: a) elevación de la ALT (alanino aminotransferasa) dos veces por encima del límite alto normal (LAN); b) aumento de la concentración de bilirrubina conjugada dos veces por encima del LAN, y c) elevación de la AST (aspartato aminotransferasa), de la FA (fosfatasa alcalina) y de la concentración total de bilirrubina, siempre que uno de los valores sea el doble del LAN. La lesión es de tipo hepatocelular (citolítica) cuando hay un aumento de la ALT dos veces por encima del LAN o cuando la relación ALT/FA es mayor de 5/1. La lesión hepática es colestásica cuando hay un aumento de FA dos veces por encima del LAN o cuando ALT/FA es menor de 2/1. La lesión mixta incluye alteraciones bioquímicas con aumento de ALT y de FA, con una relación ALT/FA mayor de 2/1 pero inferior a 5/11,5,6.
Una lesión hepática aguda es la que evoluciona en menos de tres meses; si las anomalías bioquímicas persisten durante más tiempo, la lesión se considera crónica6. La lesión hepatocelular (citolítica) por fármacos carece de características específicas, manifestándose igual que una hepatitis aguda vírica: incremento notable de las transaminasas y hallazgos en la biopsia de necrosis (predominio centrolobulillar) asociada a infiltrado inflamatorio (la presencia de eosinófilos indica la etiología tóxica). La retirada del fármaco suele comportar una rápida mejoría, con recuperación completa en uno a tres meses. Rara vez acontece una hepatitis fulminante o subfulminante, con un riesgo de mortalidad próximo al 90%. En otras ocasiones el curso de la reacción tóxica puede ser más insidioso y progresivo, evolucionando a una hepatitis crónica e, incluso, a cirrosis1,5.
La lesión colestásica se puede presentar de dos formas: a) colestasis pura (ictericia, prurito y coluria, con aumento de FA, bilirrubina conjugada y gammaglutamiltranspeptidasa, pero con transaminasas normales); se observa especialmente con los derivados hormonales y su retirada comporta una completa resolución, y b) hepatitis aguda colestásica (colestasis bioquímica asociada a dolor abdominal y fiebre, simulando una obstrucción aguda biliar); las características de hipersensibilidad son frecuentes y el pronóstico suele ser mejor que en la forma de lesión hepatocelular. En la lesión mixta existe una mezcla de las alteraciones observadas en las dos formas descritas, hepatocelular y colestásica, siendo frecuente la ictericia; el pronóstico suele ser favorable1,5.
La importancia de los fármacos como causa de hepatopatía no subyace en el total de número de casos, que es relativamente pequeño, sino en la gravedad de algunas reacciones y en su potencial reversibilidad siempre que se reconozca tempranamente la etiología tóxica. A pesar de que la hepatotoxicidad por fármacos puede manifestarse clínicamente como una hepatitis aguda, esta relación causal es infrecuente. El pronóstico a veces es peor que el de las hepatitis víricas, sobre todo si no se reconoce tempranamente la etiología tóxica y se continúa administrando el fármaco1,5.
Respecto a las hepatitis fulminantes, se ha descrito que la isoniazida, el halotano, los antidepresivos y los antiinflamatorios no esteroides (AINE) son los agentes causales más comunes. El fallo hepático fulminante por fármacos parece tener mejor pronóstico que el debido a virus, pero los datos pueden estar sesgados por la mayor proporción de casos atribuibles al paracetamol7; esta toxicidad ocurre sobre todo en personas jóvenes y rara vez es fatal, aun cuando exista un daño hepático grave. Las tasas de mortalidad global por reacciones hepatotóxicas de etiología farmacológica son muy variables y pueden oscilar del 5 al 50% según las series1,5,6.
Se considera que los medicamentos son una causa poco frecuente de hepatitis crónica, alrededor del 1%, a la vez que las series sobre trasplante hepático apenas incluyen a pacientes con hepatopatías crónicas de etiología tóxica en estadio final1,8. Por el contrario, los fármacos son la causa más frecuente de enfermedad granulomatosa hepática aguda; también se considera que son el principal factor etiológico de alteraciones vasculares hepáticas, incluyendo peliosis hepatis, enfermedad venooclusiva, hipertensión portal no cirrótica e hiperplasia nodular regenerativa1,5.
Factores de riesgo de hepatotoxicidad
Su conocimiento e identificación temprana son determinantes para prevenir las reacciones tóxicas. La susceptibilidad individual depende de múltiples factores genéticos que alteran la metabolización de los fármacos (tabla 1).

Factores genéticos
Determinan la actividad de las vías metabólicas, la efectividad de los factores protectores del organismo y la regulación de la respuesta inmune. La base genética puede condicionar diferencias étnicas en cuanto a susceptibilidad para determinados fármacos, así como la afectación de varios miembros de una misma familia5,9. La existencia de polimorfismos genéticos de los CYP y enzimas hepáticas10 determina una variabilidad individual en el metabolismo de los fármacos que puede conducir a la aparición de hepatotoxicidad (tabla 2).

Edad
La incidencia de reacciones adversas es de dos a tres veces más elevada en los ancianos y se atribuye a la prescripción más frecuente, al uso concomitante de múltiples fármacos y a los efectos indirectos de enfermedades intercurrentes. El riesgo se asocia a reacciones dependientes de la dosis, debido a alteraciones en la biodisponibilidad y en el metabolismo de los fármacos. Además, la gravedad de la lesión hepática por medicamentos también parece aumentar con la edad1,5,13. La hepatotoxicidad es mucho más rara en niños, ya que metabolizan los fármacos con mayor rapidez debido al incremento de las vías metabólicas oxidativas, aunque presentan una susceptibilidad aumentada a los salicilatos (la aspirina incrementa el riesgo de síndrome de Reye en niños de 2 a 10 años)1,5.
Sexo
La frecuencia de hepatotoxicidad con expresión clínica de hepatitis aguda es mayor en las mujeres, así como la susceptibilidad para los fármacos que causan hepatitis crónica activa (oxifenisatina y alfametildopa, nitrofurantoína). El riesgo es mayor en mujeres en edad próxima a la menopausia (predominio en esta edad de la hepatitis autoinmune), y se conjetura que los factores hormonales podrían modular el sistema inmune en el sentido de predisponer a sufrir hepatotoxicidad1,5. En el sexo masculino son más frecuentes las alteraciones vasculares de la azatioprina, especialmente tras trasplante renal14, y las reacciones por hepatotóxicos ocupacionales.
Estado nutricional
La obesidad predispone al daño hepático por halotano, ya sea por su almacenamiento prolongado en el tejido adiposo o por un incremento de la actividad del CYP que lo metaboliza15. El ayuno aumenta la actividad del CYP2E1 a la vez que reduce los valores de glutatión, y ambos factores parecen contribuir al mayor riesgo de hepatotoxicidad por paracetamol en alcohólicos16.
Antecedentes de reacción adversa a fármacos
Aumentan el riesgo de reacción a otros fármacos, explicándose por una posible predisposición genética a sufrir una respuesta inmunológica; también pueden existir reacciones cruzadas entre medicamentos de la misma familia1,5.
Enfermedades concomitantes
El hiper o el hipotiroidismo se asocian a posibles alteraciones del aclaramiento hepático de los fármacos, habiéndose descrito también mayor riesgo de hepatotoxicidad en los diabéticos16. Los enfermos con lupus eritematoso sistémico (LES) o artritis reumatoide juvenil tienen una predisposición especial a sufrir hepatotoxicidad por salicilatos, al igual que está aumentado el riesgo para distintos medicamentos en pacientes con enfermedad inflamatoria intestinal o con insuficiencia renal5,16. Las alteraciones del sistema inmune pueden constituir un factor predisponente, como es el caso de la reacción por hipersensibilidad que induce el cotrimoxazol en pacientes con infección por el virus de la inmunodeficiencia humana (VIH)17.
Hepatopatía previa
Interfiere en el metabolismo de muchos medicamentos o altera su farmacodinámica18. Sin embargo, una reacción hepatotóxica es más difícil de identificar en estos casos, cuando el deterioro de la función hepática puede ser un fenómeno evolutivo de la enfermedad de base. La hepatotoxicidad puede comportar una gravedad desproporcionada en los pacientes con cirrosis, hecho que debe tenerse en cuenta al hacer una prescripción; así, la asociación amoxicilina-ácido clavulánico no será de primera elección en pacientes con insuficiencia hepática crónica, sobre todo si son de edad avanzada, por el riesgo de sufrir una hepatitis colestásica grave19. El paracetamol es el analgésico de elección en los enfermos con hepatopatía crónica, ya que en su mayor parte se metaboliza por glucuronoconjugación; sin embargo, en los alcohólicos puede aparecer toxicidad con dosis terapéuticas debido a la inducción del CYP2E1 por el alcohol, generándose un exceso de metabolito tóxico NAPQI a la vez que se reducen las reservas de glutatión necesarias para su inactivación16,20,21.
Tratamientos intercurrentes
El riesgo de hepatotoxicidad está claramente relacionado con la polimedicación, especialmente en ancianos. La hepatotoxicidad por valproato se incrementa con la ingestión previa de fármacos inductores del CYP, como fenitoína y fenobarbital. La toxicidad vascular hepática de agentes alquilantes anticancerosos se potencia con otros quimioterápicos, y el riesgo de enfermedad venooclusiva por estos tratamientos es mayor si se aplica radioterapia previamente5,18,22.
Mecanismos patogénicos de la hepatotoxicidad
Los fármacos que generan lesión hepática pueden actuar como: a) hepatotoxinas intrínsecas, es decir, produciendo toxicidad predecible, dependiente de la dosis y reproducible, o b) hepatotoxinas idiosincrásicas, originándose toxicidad no predecible, no dependiente de la dosis ni reproducible5 (tabla 3).

Hepatotoxinas intrínsecas
Suelen causar lesión a través de sus metabolitos tóxicos23, que pueden ser radicales libres (producen peroxidación de lípidos de membrana), moléculas electrofílicas (se unen covalentemente con proteínas del hepatocito) u oxígeno activo (también produce peroxidación). La forma se origina por un mecanismo directo o indirecto5,16,23: a) la hepatotoxicidad directa implica una alteración de la membrana celular y de las organelas a través de la peroxidación de lípidos de membrana, causando la necrosis hepatocitaria. El daño es usualmente citolítico, pero puede ser colestásico si la necrosis afecta principalmente al epitelio biliar. El tetracloruro de carbono actúa (necrosis zonal 3 y esteatosis) por este mecanismo, y b) las hepatotoxinas que producen daño por un mecanismo indirecto ocasionan distorsión de moléculas esenciales o el bloqueo selectivo de vías metabólicas fundamentales para la integridad celular. El daño hepático puede ser citotóxico (necrosis o esteatosis) o colestásico (interferencia selectiva con la excrección de sustancias a los canalículos biliares).
Hepatotoxinas idiosincrásicas
Producen lesión hepática de forma impredecible, no ligada a la dosis5,16. Afecta tan sólo a una pequeña proporción de pacientes tratados y puede estar mediada por idiosincrasia inmunológica o metabólica (tabla 4). La inmunológica depende de la hipersensibilidad tras un período de exposición fijo de una a 5 semanas. Se suele asociar a fiebre, eritema cutáneo y eosinofilia, así como a linfocitosis con formas atípicas. En la biopsia hepática se aprecia un infiltrado inflamatorio granulomatoso con abundantes eosinófilos. La reexposición al fármaco condiciona una recidiva temprana. En cuanto a la metabólica, no se asocia a manifestaciones de hipersensibilidad. Tampoco se reproduce tempranamente tras la reexposición, ya que depende de una acumulación determinada de metabolito tóxico.

Tipos celulares que pueden sufrir una lesión hepatotóxica
El hepatocito es la célula diana principal del efecto tóxico de los medicamentos, pero cualquier célula hepática (epitelio ductal, endotelio, células de Ito) puede resultar afectada de forma aislada o conjuntamente con otras, originando distintos síndromes hepáticos agudos o crónicos11 (tabla 5).

Lesiones hepáticas agudas
Los agentes tóxicos, en general, producen degeneración o necrosis hepatocitaria (daño citotóxico) o interfieren con el flujo de bilis (daño colestásico)1,5,10,16.
La expresión morfológica del daño citotóxico es doble: b) necrosis, que puede ser zonal (afecta al lobulillo, pero conservando su estructura) o difusa (desestructuración del lobulillo). La necrosis zonal se diferencia en centrolobulillar (zona 3), lesión típica del paracetamol, tetracloruro de carbono y halotano; periférica (zona 1), o mediozonal (zona 2). La necrosis difusa puede tener un patrón similar al de la hepatitis vírica (p. ej., por metildopa). Las transaminasas se elevan de 8 a 500 veces por encima del LAN, reflejando cuatro formas clínicas distintas: hipertransaminasemia sin síntomas, hepatitis anictérica, hepatitis ictérica o hepatitis fulminante24, y b) esteatosis, que puede ser microvesicular (tetracicilina, valproato) o macrovesicular (etanol, metotrexato). Habitualmente no altera las pruebas de función hepática ni suele tener expresión clínica, aunque la aparición de astenia, fiebre, ictericia, ascitis o edemas puede indicar daño hepático irreversible. Los hallazgos en la biopsia son similares a los del hígado graso del embarazo o a los del síndrome de Reye5,16,24.
También se han descrito dos síndromes de daño colestásico: la colestasis aguda, tipo canalicular, asociada a esteroides sexuales, y la colestasis hepatocanalicular, con hepatitis, como la causada por clorpromazina. Clínica y bioquímicamente son similares a la ictericia obstructiva extrahepática. En la colestasis pura la lesión es de predominio centrolobulillar, donde se aprecian depósitos de bilirrubina en los hepatocitos y dilatación de los canalículos biliares que contienen pigmento biliar; en la hepatitis colestásica se asocia, además, un infiltrado inflamatorio en los tractos portales24,25.
Lesión hepática crónica
Puede ser el resultado de una exposición prolongada o bien la secuela de una lesión aguda. En la tabla 6 se detallan los diferentes tipos de lesión y los principales fármacos que las causan1,5,11,16.

Diagnóstico de la hepatotoxicidad por fármacos
Es un reto difícil en la práctica clínica diaria11,26. Se considera que la lesión hepática por fármacos está infradiagnosticada por el bajo nivel de sospecha clínica y porque el diagnóstico diferencial es habitualmente problemático (tabla 7). Se considera que es de importancia capital hacer un diagnóstico preciso y temprano, incluyendo la identificación del fármaco causante en caso de que más de uno esté bajo sospecha, ya que se puede prevenir la evolución a hepatopatías más graves o crónicas (incluyendo fallo hepático fulminante) y evitar la recurrencia.

Los objetivos clínicos se centran en caracterizar que una reacción adversa a fármacos es la causa de una determinada lesión y en identificar los agentes causantes, y la recuperación se facilita al interrumpir el tratamiento1,5,16. La ausencia de pruebas diagnósticas específicas, junto al hecho de que la reexposición dirigida rara vez está justificada por constituir un riesgo inaceptable (particularmente en hepatitis inmunoalérgicas) y originar posibles resultados falsos negativos (idiosincrasia metabólica), representa un problema para alcanzar un diagnóstico de certeza. Es decisivo que los médicos generales tomen conciencia respecto a la necesidad de considerar siempre la posibilidad de daño hepático por fármacos y tóxicos, remitiendo a continuación los datos a especialistas que conozcan los factores determinantes para establecer un diagnóstico de certeza27. Es preceptiva la obtención de una historia clínica dirigida, teniendo en cuenta que cualquier medicamento puede potencialmente causar daño hepático.
La implicación etiológica de un fármaco dependerá de varios factores6,11: a) hacer una evaluación semicuantitativa para definir las reacciones como indicativas de, compatibles con o incompatibles con hepatotoxicidad; b) definir los criterios cronológicos con implicación diagnóstica, como el tiempo de inicio de la reacción (considerado indicativo si es entre una semana y tres meses; compatible si es entre tres meses y un año, e improbable si es mayor de un año), el curso tras la supresión del tóxico (la hepatitis aguda suele recuperarse en pocas semanas, mientras que las hepatitis colestásicas pueden prolongarse más de 6) y la respuesta a la readministración de la hepatotoxina sospechosa; c) determinar los signos y síntomas, aunque inespecíficos, que apunten a la implicación de un fármaco, así como los hallazgos sistémicos (fiebre, erupción cutánea y eosinofilia) y la positividad de ciertas pruebas serológicas específicas (anticuerpos antimitocondriales tipo 6 para la iproniazida, anti-CYP 1A2 para la dihidralacina o anti-CYP 2E1 para el halotano), y d) considerar la existencia de factores que implican mayor riesgo de hepatotoxicidad: edad superior a 50 años, ingestión de múltiples fármacos o el uso de un medicamento con conocido efecto hepatotóxico. Además, es preciso la exclusión de otras posibles causas de enfermedad hepática (tabla 8).

La biopsia hepática puede estar indicada en determinados casos con dificultad diagnóstica y, especialmente, tras la retirada del fármaco sospechoso. Algunos hallazgos histológicos indican una posible etiología por medicamentos (tabla 9). También es interesante realizar una biposia hepática en el caso de fármacos de difícil sustitución para el paciente (amiodarona), por la posibilidad de excluir su implicación lesiva.

Una reexposición positiva, con recurrencia del daño hepático, es la evidencia definitiva de que un medicamento causó dicha lesión. Sin embargo, la reexposición deliberada tiene unas implicaciones éticas, dado el riesgo de morbimortalidad, que obligan a evitarla sobre todo cuando la hepatotoxidad es grave28; podría considerarse en el caso de que el fármaco fuera esencial para el paciente o si es la primera vez que se considera implicado en una reacción de hepatotoxicidad.
Los análisis serológicos para detectar anticuerpos inducidos por un fármaco constituyen pruebas específicas, pero rara vez son positivas en las reacciones hepáticas por medicamentos y tan sólo cuando están mediadas inmunológicamente29. En la hepatotoxicidad por idiosincrasia metabólica existen pruebas capaces de detectar deficiencia del sistema enzimático detoxificador30. No obstante, los resultados no siempre son aplicables en la práctica clínica y se requieren más estudios en este sentido.
La posibilidad de que exista hepatotoxicidad es alta en el caso de hepatitis aguda o colestasis que no se explican por etiologías comunes, sobre todo en ancianos31, tras la introducción reciente de un nuevo fármaco32 o cuando se aprecia una combinación atípica de hallazgos patológicos. Las limitaciones diagnósticas han llegado a plantear nuevas estrategias de vigilancia continuada que permitan generar señales y estimar riesgos como, por ejemplo, la estrategia de análisis de casos y controles de enfermedades infrecuentes pero graves, ya que a menudo son inducidas por fármacos33. En este tipo de investigación epidemiológica analítica los sujetos se seleccionan según padezcan (casos) o no (controles) una enfermedad particular objeto de estudio y se comparan con respecto a la proporción de aquellos que tienen una historia de exposición a fármacos o a unas determinadas características de interés. No obstante, una de sus principales limitaciones es su escaso valor para estudiar los riesgos asociados con fármacos de indicaciones y uso reducidos.
Hepatotoxicidad por fármacos indicados en patología reumática
Los AINE son los fármacos más utilizados para el tratamiento de una gran variedad de problemas inflamatorios agudos o crónicos32; además, también son los que se prescriben con mayor frecuencia en el mundo. A pesar de ello, la incidencia de lesión hepática por AINE es relativamente baja, estimándose en menos del 0,1%34,35. Está bien documentada para el sulindaco y el diclofenaco, con un espectro clínico que va desde elevaciones transitorias y leves de las transaminasas hasta fallo hepático fulminante como los casos fatales por benoxaprofén36. El riesgo de daño hepático aumenta con la ingestión simultánea de otra medicación hepatotóxica37. Se ha descrito un leve aumento de las transaminasas hasta en el 25% de los pacientes con artritis reumatoide o con LES que reciben tratamiento34,38.
La hepatotoxicidad por sulindaco está bien documentada, comprobándose tras la reexposición39. Se han reconocido los dos patrones típicos de daño hepático, citolítico y colestático, pero este último es el más común. La lesión aparece típicamente a las 8 semanas de iniciado el tratamiento y el mecanismo de la hepatotoxicidad parece ser por hipersensibilidad; los casos fatales son debidos a fallo hepático fulminante por posible hipersensibilidad intensa, y aproximadamente el 75% son mujeres casi siempre mayores de 50 años40.
El diclofenaco provoca lesión hepática de tipo mixto y hepatocelular por un mecanismo idiosincrásico, probablemente secundario al metabolismo del fármaco, pues la inhibición del CYP2C reduce el daño citolítico41. Aunque un 15% de los pacientes tratados desarrollan una hipertransaminasemia de hasta tres veces el LAN, se estima que una hepatotoxicidad manifiesta ocurre en uno a 5 de cada 100.000 exposiciones, pareciendo más susceptibles las mujeres y los pacientes con osteoartritis42,43. En ocasiones puede producir una lesión hepatocelular crónica, similar a la de la hepatitis autoinmune44. La incidencia de hepatotoxicidad por indometacina parece ser baja y la lesión puede ser hepatocelular o colestásica, habiéndose descrito la posibilidad de casos de hepatitis fulminante45.
El ibuprofeno no parece ser un fármaco tóxico a dosis bajas. En los pocos casos descritos el daño suele presentar un patrón mixto, hepatocelular y colestásico. No está claro el mecanismo patogénico de la hepatotoxicidad, aunque la asociación con el síndrome de Stevens-Johnson apunta a una reacción de tipo inmunológico5,16,46. El mecanismo de hepatotoxicidad del naproxeno es desconocido, siendo una causa rara de lesión hepática y no existiendo evidencia de mortalidad. En los pacientes que presentaron ictericia, el diagnóstico se estableció varios meses depués de haber iniciado el tratamiento y el síndrome ictérico se resolvió rápidamente tras la retirada del fármaco47. La mayoría de los casos de hepatotoxicidad secundaria a fenilbutazona presentaban signos de daño hepatocelular similar al de una hepatitis vírica, con un patrón colestásico en una minoría de pacientes; la patogenia de la lesión parece mediada inmunológicamente, apreciándose granulomas en un tercio de las biopsias hepáticas46,48.
El benoxaprofén fue retirado en EE.UU. por provocar casos fatales de lesión hepática, la mayoría de las veces en mujeres de edad avanzada, con daño predominantemente colestásico y fallo renal asociado; también fueron retirados por su inaceptable grado de hepatotoxicidad el cincofeno e ibufenaco5,16,46. Un hecho similar ocurrió con el droxicam, al demostrarse que causaba hepatitis colestásica por un mecanismo de hipersensibilidad49. Igualmente se han comunicado varios casos de fallo hepático fulminante, con indicación de trasplante hepático o con fallecimiento, en relación con el bronfenaco, por lo que se procedió a retirarlo del mercado pocos meses después50. También se han descrito casos de hepatotoxicidad secundaria a piroxicam, oxaprozín, tolmetín, etodolaco y ácido mefenámico5,16,46. Por el contrario, las reacciones hepatotóxicas por ketorolaco, meclofenamato o tramadol constituyen una rareza.
Los salicilatos pueden provocar una necrosis hepática focal que suele ser dependiente de la dosis, en especial cuando los valores hemáticos sobrepasan los 25 mg/dl. Parece ser más frecuente en niños con artritis reumatoide juvenil y fiebre reumática aguda, así como en adultos con artritis reumatoide y LES. La lesión se resuelve rápidamente tras la suspensión del fármaco5,16,51. La administración de aspirina en el contexto de una enfermedad vírica puede desencadenar un síndrome de Reye16,24. El diflunisal, un derivado difluorofenilo, ha sido implicado en hepatotoxicidad probablemente por idiosincrasia inmunológica38. La sulfasalacina y la mesalazina, de estrucutra química similar a los salicilatos, también se han relacionado con hepatotoxicidad, presentando manifestaciones inmunológicas asociadas52. La nimesulida, un inhibidor selectivo de la enzima COX-2, puede provocar daño hepático hepatocelular o colestásico por idiosincrasia metabólica o inmunológica53, esta última en probable relación con hipersensibilidad al grupo funcional sulfonanilida similar a la descrita para la dapsona o la sulfapiridina54.
Las estrategias para prevenir la hepatotoxicidad por AINE pasan por: a) evitar su indicación conjunta con otros fármacos potencialmente hepatotóxicos; b) utilizarlos con precaución en aquellos pacientes con enfermedad hepática crónica o en los consumidores de alcohol en exceso, y c) prescribir el preparado con menor actividad hepatotóxica. En los pacientes de riesgo se recomienda la monitorización mensual de la función hepática. Parece razonable informar al enfermo de la posibilidad de daño hepático, así como de los signos y síntomas de alarma, cesando el tratamiento inmediatamente en caso de que surgiera o se detectara algún indicio de hepatotoxicidad.
El metotrexato es un fármaco que requiere una atención especial al considerar su capacidad hepatótoxica, debido a su implicación en el desarrollo gradual y silente de cirrosis. El metotrexato está indicado en pacientes con psoriasis grave y con artritis reumatoide, y se le supone el riesgo de causar hepatotoxicidad tras un período de varios años sin que se aprecien síntomas o anomalías bioquímicas indicativos de enfermedad hepática55,56. El metotrexato se ha asociado con el desarrollo de esteatosis, fibrosis progresiva y cirrosis57. La intensidad del daño hepático guarda una relación directa con la duración del tratamiento, a la vez que se relaciona de forma inversa con la duración del intervalo entre las dosis, de forma que la incidencia de hepatopatía tóxica es muy baja en los pacientes que reciben una dosis acumulada inferior a 2 g58,59.
Los pacientes con psoriasis parecen más susceptibles a sufrir hepatotoxicidad por metotrexato que aquellos con artritis reumatoide, aunque los principales factores de riesgo son: edad avanzada, diabetes, obesidad, alcoholismo activo, hepatopatía previa, insuficiencia renal crónica y dosis total acumulada59-61. El metotrexato está contraindicado en los enfermos que tengan una hepatitis aguda o crónica con actividad, cirrosis descompensada, insuficiencia renal o alcoholismo activo. En los pacientes con alguno de estos factores de riesgo es preciso realizar una biopsia hepática al inicio del tratamiento y posteriormente durante el seguimiento57,61. El desarrollo de cirrosis es silente, sin manifestaciones clínicas y bioquímicas, y la biopsia es el único método seguro para establecer un diagnóstico de certeza. La importancia radica en que la fibrosis tiende a regenerar cuando se suspende el tratamiento con metotrexato56,61.
Aunque los protocolos clínicos varían según los grupos, la tendencia más generalizada aconseja no realizar biopsia en los pacientes con bioquímica hepática normal y sin factores de riesgo, indicando tan sólo el estudio histológico en caso contrario. La pauta no está perfectamente establecida, pero se aconseja obtener una biopsia basal en los pacientes de riesgo y repetir el control histológico cada 2-4 años o antes si se detectan alteraciones bioquímicas significativas y persistentes57. No obstante, el riesgo de hepatotoxicidad por metotrexato es bajo, sobre todo si el paciente se vuelve abstemio, se excluyen del tratamiento a los enfermos con factores de riesgo graves o se administran dosis muy bajas. Las normas del Colegio Americano de Gastroenterología para el tratamiento con metotrexato se resumen en la tabla 1062,63.

La leflunomida es nuevo fármaco con acción antiinflamatoria y antiproliferativa recientemente aprobado (septiembre de 1999) para el tratamiento de la artritis reumatoide. Se incluye en el grupo denominado con el acrónimo del inglés DMARD (fármacos modificadores de la enfermedad)64,65. Se calcula que han sido tratados más de 200.000 pacientes en todo el mundo, con un aceptable perfil de seguridad establecido por diferentes estudios. Los efectos adversos más comunes fueron diarrea, dispepsia, eritema cutáneo, alopecia, hipertensión y elevación de las enzimas hepáticas66-68. Sin embargo, desde su aprobación se ha comprobado que es un tratamiento eficaz para muchos pacientes con artritis reumatoide y que es aceptablemente bien tolerado.
Respecto a las alteraciones hepáticas, se han notificado 295 casos entre un número total de 104.000 pacientes tratados por año. Las reacciones hepatotóxicas aparecieron en el transcurso de los primeros 6 meses de tratamiento y fueron graves en 121 casos, incluyendo dos cirrosis hepáticas y 15 pacientes con insuficiencia hepática aguda grave que se asoció a fallecimiento en 9 de ellos. No obstante, los hechos son un tanto confusos en el sentido de que 101 pacientes (78%) que habían presentado efectos adversos graves recibían simultáneamente con leflunomida otros medicamentos con capacidad potencial de inducir hepatotoxicidad; en concreto, el 58% de los pacientes con alteraciones de la bioquímica hepática habían recibido concomitantemente metotrexato o AINE. Además, en 33 enfermos (27%) que presentaron hepatotoxicidad grave se demostró la existencia de otros factores de riesgo como alcoholismo activo, hepatopatía previa (hepatitis crónica vírica B o C) insuficiencia cardíaca aguda, neumopatía grave o carcinoma de páncreas66-68. Un análisis preliminar del perfil de prescripción de la leflunomida apunta a que las recomendaciones sobre la monitorización de la función hepática y los procedimientos de eliminación rápida del fármaco (lavados) podrían no haberse respetado de una manera estricta.
Las distintas reacciones graves mencionadas no pueden atribuirse a la leflunomida con absoluta certeza, pero tampoco es posible excluir una relación causal. Así pues, considerando esta posibilidad, la Agencia Española del Medicamento destaca que la leflunomida está contraindicada en pacientes con trastornos de la función hepática y estima que es esencial que se respeten las recomendaciones para el control de la función hepatocelular de los enfermos susceptibles de tratamiento (tabla 11). Si aparece algún efecto adverso grave o es necesario eliminar rápidamente el metabolito activo de la leflunomida (embarazo, sustitución por otro DMARD), deben cumplirse las recomendaciones para el seguimiento y procedimiento de lavado (tabla 11).

No se recomienda el tratamiento concomitante con metotrexato o con otros fármacos potencialmente hepatotóxicos, ya que aumenta el riesgo de reacciones adversas graves. Sin embargo, la combinación terapéutica de leflunomida con metotrexato no aparece como contraindicada. En dos estudios recientes69,70 se ha llegado a la conclusión de que los mecanismos de acción complementarios justifican la indicación conjunta de ambos fármacos y que esta combinación puede ser eficaz para el tratamiento de los pacientes que no han respondido al metotrexato como monoterapia. Desde el punto de vista de la seguridad del tratamiento combinado, se apreció un incremento de los valores séricos de las enzimas hepáticas por encima de 2 veces el LAN en el 6,2% de los pacientes tratados, frente al 1,5% en el grupo que recibió metotrexato más placebo, aunque sin evidencia de hepatotoxicidad grave. Además, la reducción de la dosis de leflunomida se asoció con frecuencia a la normalización de las transaminasas.
La impresión práctica es que el perfil beneficio-riesgo de la combinación terapéutica es favorable, siempre que se haga una selección estricta para excluir a los pacientes de riesgo, se cumplan las recomendaciones para el control de la función hepatocelular y se proceda a reducir las dosis en función de la eficacia y tolerancia al tratamiento combinado. En la actualidad se está realizando un ensayo clínico aleatorio sobre la eficacia y seguridad de la combinación de ambos fármacos, cuyos resultados podrán aclarar muchos de los aspectos aún en debate.
Tratamiento de la hepatopatía por fármacos
Se puede afirmar que, en general, el tratamiento de la lesión hepática producida por fármacos es insatisfactorio. La excepción es el paracetamol, para el que se dispone de un antídoto. La medida más importante consiste en la suspensión inmediata del fármaco sospechoso de producir daño hepático. Puede resultar problemático cuando se administran varios medicamentos a la vez; en estos casos, la norma se basa en considerar al que se haya introducido más recientemente como el posible agente etiológico o, en su defecto, a aquel o aquellos que previamente hayan sido reconocidos como potencialmente hepatotóxicos1,5,6,16.
Si el medicamento es una hepatotoxina dependiente de la dosis (metales, paracetamol), se tomarán las medidas oportunas para su eliminación del organismo en relación con el tiempo transcurrido desde la toma del fármaco. En algunos casos está indicada la utilización de antídotos específicos, como la N-acetilcisteína en la intoxicación por paracetamol, y los quelantes como la desferroxamina en el caso del hierro y el dimercaprol6,16,71.
Se plantea la posibilidad de que la utilización de agentes hepatoprotectores puede ser una medida importante en un futuro. Es el caso de la N-acetilcisteína, aun cuando hayan transcurrido más de 16 h tras la ingestión de paracetamol55, ya que se ha indicado que podría mejorar la microcirculación hepática, y de la prostaglandina E y prostaciclinas sintéticas. Los corticoides están indicados en casos individuales y obtienen cierta eficacia en la hepatitis granulomatosa por alopurinol, en la hepatitis crónica por etretinato en la inducida por diclofenaco6,16,72. La impresión general es que los corticoides no están indicados en las hepatitis agudas o crónicas graves ni en las colestasis inducidas por fármacos6,16,73.
Cuando la reacción tóxica es intensa y se acompaña de síntomas graves, puede ser necesario el ingreso hospitalario del paciente y la aplicación de un tratamiento de soporte adecuado. Se ha de prestar especial atención a un correcto balance hidroelectrolítico y nutricional, tratando los trastornos de la coagulación con vitamina K o plasma fresco y concentrados de factores si es preciso. Para el prurito, síntoma principal en caso de colestasis, el tratamiento de elección es la colestiramina, aunque a veces la rifampicina ha demostrado su eficacia74,75. Recientemente se ha propuesto la administración de ácido ursodesoxicólico para las colangitis crónicas de larga evolución; no obstante, dado el pequeño número de casos, es difícil demostrar un efecto beneficioso evidente11,58. Es igualmente útil la monitorización de los factores V y VII de la coagulación. Ante el deterioro progresivo de la función hepática con fallo fulminante o subfulminante, está indicado el trasplante hepático, con muy buenos resultados si no se demora excesivamente la obtención de un donante.