





El deterioro funcional de los órganos y sistemas es la manifestación principal del envejecimiento. En el caso del hueso, se considera que el envejecimiento constituye un factor de riesgo primordial en la pérdida de masa y calidad ósea, lo que trae como consecuencia un aumento de la incidencia de fracturas. Múltiples factores parecen contribuir a esta situación, entre los que se pueden incluir los cambios metabólicos que tienen lugar en el propio tejido óseo al envejecer. La mayoría de los conocimientos actuales sobre los mecanismos asociados a la osteopenia/osteoporosis durante el envejecimiento se han generado a partir de estudios en modelos animales (fundamentalmente, ratas y ratones) y en cultivos celulares obtenidos de sujetos de distinta edad. En este trabajo, hemos revisado y compendiado estos estudios, que han permitido establecer las bases fisiológicas y moleculares que subyacen en las alteraciones óseas asociadas al envejecimiento.
Deterioration of organ and systems function are the principal signs of aging. Aging is also believed to be a major factor in the loss of bone mass and quality, which in turn leads to an increase in the risk of fractures. Several factors seem to contribute to this scenario, with metabolic changes related to aging in the bone tissue itself being among them. Most of the current knowledge on the mechanisms associated with osteopenia/osteoporosis during aging has been generated from research in animal models (mainly rats and mice) and cell cultures derived from subjects of different ages. In this work, we have reviewed and summarised these studies, which have begun to establish the physiological and molecular basis of the bone alterations related to aging.
En el envejecimiento tiene lugar un deterioro funcional de los tejidos, órganos y sistemas del organismo, especialmente de aquellos que mantienen la homeostasis corporal y, consecuentemente, la salud del individuo, lo que supone una mayor morbilidad y mortalidad con la edad1. Estudios epidemiológicos han demostrado una pérdida de masa ósea a partir de la tercera década de la vida, tanto en hombres como en mujeres, momento en el que se alcanza el pico máximo de dicha masa ósea2,3. En un principio, esta pérdida de masa ósea va a afectar principalmente al hueso trabecular, pero en una fase posterior del proceso de envejecimiento también afecta al hueso cortical, siendo este hecho independiente de los cambios que tengan lugar en las hormonas sexuales2–4. No obstante, tras la menopausia, la osteopenia se acelera como consecuencia de la pérdida del papel protector de los estrógenos5.
Actualmente, se considera que el envejecimiento constituye un factor de riesgo primordial en la pérdida de masa y de resistencia ósea, que lleva como consecuencia un aumento de la incidencia de fracturas. Es más, la edad per se puede contribuir al aumento del riesgo de sufrir una fractura en relación con múltiples factores; algunos extraóseos, como las alteraciones neuromusculares relacionadas con la inmovilidad, el exceso de glucocorticoides y la insuficiencia renal (causante de un hiperparatiroidismo secundario debido al déficit de calcitriol)6. También hay que considerar en este contexto los cambios metabólicos que tienen lugar con la edad en el propio tejido óseo, como: 1) la peor calidad de las fibras de colágeno7; 2) la menor capacidad de reparación de microfracturas8; y 3) el aumento relativo de resorción ósea frente a la formación ósea9.
Cambios histológicos en el tejido óseo con la edadLos modelos en roedores (ratones y ratas) son una poderosa herramienta para la investigación de diversas afecciones humanas, y la osteopenia/osteoporosis asociada al envejecimiento no constituye una excepción. Al igual que sucede en los seres humanos, las ratas sufren una pérdida de masa ósea con la edad10. Además, la mineralización del osteoide tras la ablación medular en el fémur disminuye de forma significativa de los 6 a los 24 meses de edad (de la edad adulta a la vejez) en ratas Wistar macho11. Esta respuesta disminuida de la regeneración ósea asociada a la edad se refleja también en las propiedades histomorfométricas y estructurales del hueso12. La pérdida de masa ósea con la edad, tanto en el hueso cortical como en el trabecular, depende de la estirpe de rata estudiada10,13. Además, estudios en ratas envejecidas demuestran un aumento en la relación osteoclastos/osteoblastos asociado a la pérdida de hueso trabecular14.
Así mismo, tanto en ratones no-consanguíneos15–17 como en cepas consanguíneas18–22 se han observado cambios en la masa ósea asociados a la edad. Los ratones no-consanguíneos de la cepa CW-1 desarrollan su pico de masa ósea a los 12, 15 y 18 meses de edad en el hueso cortical, trabecular y subcondral, respectivamente, y sufren una pérdida de masa ósea cortical y trabecular entre un 20 y un 60%, respectivamente, hasta la etapa final del proceso de envejecimiento (alrededor de 30 meses)15,17. Esta pérdida de masa ósea se relaciona en estos animales con una insuficiente actividad osteoblástica y con una resorción ósea aumentada16,17. En ratones consanguíneos, de las cepas C57BL/6J, C3H/HeJ, BALB/cByJ y DBA/2J, la masa ósea está regulada por factores genéticos18,23–25, observándose una disminución de la masa ósea a partir de los 12 meses de edad18. De manera similar a lo que ocurre en las hembras CW-1, ratones macho de la cepa C57BL/6J pierden el 60% del volumen óseo trabecular y el 12% del espesor trabecular en la tibia entre los 6 y los 24 meses de edad, a la vez que disminuye la masa ósea total un 10%26. Utilizando cultivos primarios de células de ratones viejos (24 meses) C57BL/6 y BALB/c, se observó un moderado aumento en el número total de células de la médula ósea, de acuerdo con un aumento del tamaño cavitario27. El ratón macho de la estirpe C57BL/6 alcanza la madurez a las 12 semanas, no habiendo cambios significativos en la densidad mineral ósea ni en las propiedades mecánicas del fémur hasta las 42 semanas. Sin embargo, entre las 4 y las 22 semanas, descienden tanto la velocidad de formación ósea, como la tasa de aposición mineral y la superficie de hueso erosionada28.
El tipo de patrón de osteopenia en ratones10,23 se observa también en los seres humanos, donde existe una pérdida lineal de hueso trabecular con la edad, mucho mayor que en el hueso cortical3,29–31. El área cortical de la diáfisis femoral aumenta hasta los 70 años, pero después decae tanto en mujeres como en varones; mientras que entre los 21 y 97 años el área medular se triplica en las mujeres y se duplica en los varones32. Por otro lado, la edad se asocia a un aumento de la porosidad intracortical en el fémur32. Es de destacar, que en humanos la expansión de la cavidad medular y la disminución del hueso cortical son dos de los principales procesos relacionados con las fracturas osteoporóticas32,33.
El mantenimiento de las propiedades mecánicas del hueso es de gran importancia para evitar lesiones y fracturas. En ratones C57BL/6J se ha comprobado que con la edad se mantienen dichas propiedades mecánicas por mecanismos similares a los observados en humanos34. Este mantenimiento, se debe en parte al incremento del momento de inercia debido al aumento del diámetro del periostio, aunque con una mayor porosidad cortical observada en ratones C57BL/J6 machos de más de dos años de edad28.
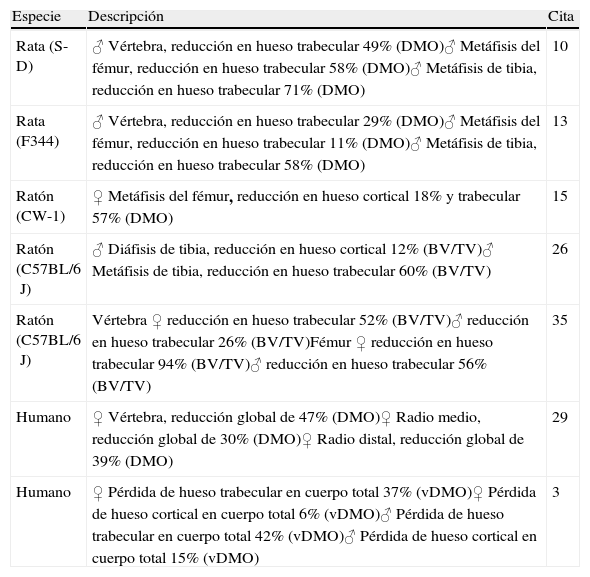
Las consecuencias del envejecimiento en el tejido óseo podrían estar matizadas por las diferencias sexuales en los modelos animales usados en el laboratorio. A este respecto, un estudio reciente en ratones de la estirpe C57BL/6J demuestra que, aunque la densidad mineral ósea (DMO) y el peso total del animal aumentan con la edad en ambos sexos, el volumen óseo trabecular disminuye a partir de las 8 semanas; siendo este descenso más pronunciado en las hembras. Además, en éstas se produce un descenso anticipado de DMO en vértebras y en el fémur distal con respecto a los machos35. Este descenso se relaciona con una disminución en el número de trabéculas y con un ligero aumento del grosor trabecular. Durante el envejecimiento, los ratones machos de esta estirpe presentan un menor número de trabéculas, junto a una disminución de la conectividad trabecular26. Como se ha mencionado, estos ratones macho llegan a perder el 60% del volumen de hueso esponjoso entre los 6 y los 24 meses, similar a lo que ocurre en humanos26. Se ha sugerido que este descenso del volumen trabecular podría estar relacionado con un descenso en la actividad física y, como consecuencia, un aumento de peso que disminuiría los estímulos mecánicos en el hueso36. La tabla 1 resume los cambios observados en la masa ósea en modelos de roedores y en humanos con la edad.
Cambios en la masa ósea con la edad en roedores y en humanos
| Especie | Descripción | Cita |
| Rata (S-D) | ♂ Vértebra, reducción en hueso trabecular 49% (DMO)♂ Metáfisis del fémur, reducción en hueso trabecular 58% (DMO)♂ Metáfisis de tibia, reducción en hueso trabecular 71% (DMO) | 10 |
| Rata (F344) | ♂ Vértebra, reducción en hueso trabecular 29% (DMO)♂ Metáfisis del fémur, reducción en hueso trabecular 11% (DMO)♂ Metáfisis de tibia, reducción en hueso trabecular 58% (DMO) | 13 |
| Ratón (CW-1) | ♀ Metáfisis del fémur, reducción en hueso cortical 18% y trabecular 57% (DMO) | 15 |
| Ratón (C57BL/6J) | ♂ Diáfisis de tibia, reducción en hueso cortical 12% (BV/TV)♂ Metáfisis de tibia, reducción en hueso trabecular 60% (BV/TV) | 26 |
| Ratón (C57BL/6J) | Vértebra ♀ reducción en hueso trabecular 52% (BV/TV)♂ reducción en hueso trabecular 26% (BV/TV)Fémur ♀ reducción en hueso trabecular 94% (BV/TV)♂ reducción en hueso trabecular 56% (BV/TV) | 35 |
| Humano | ♀ Vértebra, reducción global de 47% (DMO)♀ Radio medio, reducción global de 30% (DMO)♀ Radio distal, reducción global de 39% (DMO) | 29 |
| Humano | ♀ Pérdida de hueso trabecular en cuerpo total 37% (vDMO)♀ Pérdida de hueso cortical en cuerpo total 6% (vDMO)♂ Pérdida de hueso trabecular en cuerpo total 42% (vDMO)♂ Pérdida de hueso cortical en cuerpo total 15% (vDMO) | 3 |
BV/TV: volumen óseo/volumen tisular; DMO: densidad mineral ósea; S-D: Sprague-Dawley; vDMO: densidad mineral ósea volumétrica.
Uno de los factores que regulan la masa ósea es el cociente osteoprotegerina (OPG)/ligando del receptor activador del factor nuclear (NF)-κB (RANKL) y se ha demostrado que este cociente se ve alterado con la edad. De hecho, se ha encontrado que la relación OPG/RANKL es menor en las tibias de ratones C57BL/6 machos de 6-24 meses frente a aquéllos de un mes y medio de edad; fundamentalmente debido al aumento de la expresión del RANKL37. Estos resultados son coherentes con los observados en cultivos de osteoprogenitores de la médula ósea procedentes de sujetos de distintos grupos de edad38–40. Un cociente OPG/RANKL disminuido con la edad indicaría que habría un mayor número de osteoclastos durante el envejecimiento; de hecho, se ha demostrado la existencia de un mayor número relativo de precursores osteoclásticos respecto al de osteoblastos en la médula ósea de ratones C57BL/6 viejos41.
En ratones envejecidos C57BL/6 se ha demostrado, que paralelamente a la edad, aumenta la producción de glucocorticoides endógenos a través de la activación de la enzima 11 β-hidroxiesteroide deshidrogenasa tipo 1. Este hecho se relaciona con una reducción de la vida media de las células óseas (osteoblastos y osteoclastos), junto a la disminución del transporte de solutos a través de las conexiones canaliculares osteocíticas, así como de la angiogénesis, fundamental en el metabolismo óseo42,43. Todo ello puede deberse, al menos en parte, a que los glucocorticoides inhiben la activación del factor de hipoxia inducible-1 (HIF-1) que promueve la transcripción del factor de crecimiento del endotelio vascular (VEGF)44.
Uno de los problemas asociados a los estudios del envejecimiento en este tipo de roedores es que requieren un tiempo relativamente largo (hasta dos años) para desarrollar el modelo en el que realizar la experimentación que corrobore las hipótesis planteadas. En este sentido, los modelos de ratones envejecidos prematuramente, con características similares a los envejecidos cronológicamente, presentan una serie de ventajas. Así, los ratones de la estirpe SAMP6, caracterizados en la década de los 80 como un modelo de senescencia, presentan un grosor cortical menor en el fémur y osteopenia a los 5 meses de edad, y una mayor tendencia a sufrir fracturas a edades avanzadas45,46. Se ha demostrado que a esta edad, las vértebras de estos ratones poseen un menor número de progenitores osteoblásticos y de osteoclastos y menor masa ósea47.
En resumen, un análisis meticuloso de los efectos óseos de la edad en ratas y ratones demuestra la utilidad de estos modelos animales en el estudio de la osteopenia/osteoporosis durante el envejecimiento en humanos. En particular, el ratón presenta la ventaja de una gran disponibilidad de estirpes transgénicas. Sin embargo, hay que tener en cuenta algunas particularidades de los modelos en roedores a la hora de extrapolar los resultados obtenidos en estos modelos a la práctica clínica. Entre ellos se pueden mencionar: el continuo crecimiento óseo (modelado) a partir de la placa de crecimiento; la falta de menopausia; y la ausencia de sistema haversiano y de remodelado cortical. En este sentido, un estudio desarrollado con primates del nuevo mundo (Challitrix jacchux) envejecidos, ha demostrado la idoneidad de este tipo de simios para investigar el envejecimiento óseo48.
Alteraciones de la reparación ósea con la edadSe sabe que la tasa de reparación de una fractura disminuye a partir de los 30 años, aunque se conocen mal los mecanismos moleculares subyacentes. En la actualidad, el déficit de regeneración ósea tras una fractura en la población envejecida es un problema clínico de gran importancia49. Está bien documentado que durante el envejecimiento existe una disminución de las células osteoprogenitoras, un aumento del potencial adipogénico en la médula ósea a expensas de la condrogénesis y la osteogénesis, junto con una alteración de la competencia osteoblástica para modular la osteoclastogénesis41,50,51. Estos factores pueden influir negativamente en la reparación de fracturas. Además, también se ha observado una disminución con la edad de las células endoteliales y de los factores y las vías de señalización que las modulan, indicando que la formación de vasos sanguíneos, impedida al envejecer, afecta al proceso de reparación ósea52,53.
Cambios en la función osteoblástica con la edadLos mecanismos fisiopatológicos de la osteopenia relacionada con la edad aún no están totalmente esclarecidos. Actualmente se considera clave el déficit de formación ósea al avanzar la edad, produciéndose una disminución drástica de las superficies de formación ósea frente a las de resorción, lo que indica una falta de capacidad osteoblástica para reconstituir el hueso resorbido en las unidades de remodelado óseo14,26,54,55. En principio, este hecho se atribuye a una disminución del número de osteoprogenitores derivados de células mesenquimales de la médula ósea, a costa de otros linajes alternativos (fibroblastos, adipocitos y condrocitos) compartidos por estas células mesenquimales56. Alternativamente, el envejecimiento parece afectar al crecimiento y/o a la función de las células osteoformadoras (osteoblastos). Diversas evidencias experimentales sugieren que este último tipo de mecanismo sería el predominante para explicar el déficit de formación ósea al envejecer14,57,58.
Se ha descrito una disminución asociada a la edad en el contenido total óseo de osteocalcina en los seres humanos59, así como en la actividad del promotor del gen de osteocalcina en ratones transgénicos que lo sobreexpresan60. Además, se sabe que el aumento de la osteocalcina, tanto ósea como circulante, en respuesta al calcitriol, disminuye con la edad61. Así mismo, la respuesta de otra proteína que responde a esta hormona, la calbindina D, disminuye con la edad en el duodeno de rata62.
Teniendo en cuenta la correlación entre la formación ósea objetivada mediante histomorfometría y el crecimiento de células osteoblásticas en cultivo primario ex vivo63,64, se han utilizado estos cultivos humanos (células hOB) como correlato de la situación in vivo. De este modo, se ha descrito una disminución, relacionada con el envejecimiento, de la capacidad de proliferación celular basal y tras estimulación con varios factores de crecimiento65–67; por el contrario, con la edad, las células hOB secretan más fosfatasa alcalina65. Estos hallazgos indican una mayor maduración osteoblástica durante el envejecimiento.
Las células osteoblásticas interaccionan con una variedad de factores locales y endocrinos que actúan como moduladores del remodelado óseo68,69. Entre estos últimos, la parathormona (PTH) y el metabolito activo de la vitamina D3, 1,25(OH)2D3 o calcitriol, son importantes reguladores de la función osteoblástica. Se ha observado que la respuesta a la PTH varía en las células hOB con la edad del donante de estos cultivos67,70. En esta situación, nuestro grupo ha demostrado una disminución de la expresión (génica y proteica) de la proteína relacionada con la PTH (PTHrP), un importante modulador local de la formación ósea, asociada a la del VEGF en las células hOB durante el envejecimiento71.
La respuesta ósea al calcitriol también disminuye con la edad. Así, se ha demostrado que la acción anabólica de la vitamina D depende del tipo de hueso (trabecular o cortical) y de la edad del receptor72–74. En respuesta al calcitriol, disminuye la secreción del péptido C-terminal del procolágeno tipo I y de la fosfatasa alcalina —marcadores tempranos de maduración osteoblástica—, mientras la producción de osteocalcina (a nivel de expresión génica y de proteína) —un marcador más tardío— se atenúa en las células hOB aisladas de la cadera (un hueso de composición predominantemente cortical) de sujetos mayores de 70 años75,76. La base molecular de esta deficiente respuesta al calcitriol la ha suministrado, al menos en parte, el hecho de que los niveles del ARNm de su receptor (evaluados por RT-PCR), están disminuidos en las células hOB de estos sujetos ancianos76. De acuerdo con estos hallazgos, se ha observado una disminución similar en la respuesta al calcitriol de células hOB procedentes de pacientes con bajo remodelado óseo77, así como una resistencia a la acción de la PTH sobre la 1α-hidroxilasa renal en ratas viejas78.
Así mismo, estudios in vitro en células hOB e in vivo en roedores indican que la capacidad osteogénica de una diversidad de factores locales, como el factor transformante del crecimiento β, la proteína morfogénica del hueso-2 o el factor similar a la insulina tipo 1, disminuyen con la edad67,79. Es interesante resaltar en este contexto que se ha demostrado una disminución del contenido de este último factor, paralelo al de osteocalcina, en la cabeza femoral con la edad59.
Recientemente, se ha demostrado que el tratamiento de pacientes ancianos hipertensos y osteoporóticos con antagonistas de los receptores de angiotensina II aumenta la masa ósea80,81. Este efecto beneficioso se atribuye al efecto de estos fármacos impidiendo la acción inhibitoria de la angiotensina II sobre la expresión de osteocalcina y la actividad de fosfatasa alcalina82,83. Dichos efectos inhibitorios de la angiotensina II son probablemente debidos a que este péptido, a través del aumento de AMPc, disminuye la expresión del factor de transcripción relacionado con runt 2 que regula la expresión de los marcadores osteoblásticos citados84. Este aumento de AMPc produce a su vez el incremento de la expresión de RANKL, favoreciendo así la diferenciación osteoclástica85.
Un importante modulador del envejecimiento celular es el producto del gen Klotho86. Ratones carentes de este gen sufren un envejecimiento acelerado acompañado de osteopenia. Esta osteopenia se caracteriza por una reducción del grosor cortical en fémur, tibia y vértebras, en porcentajes que varían de 20 al 40%, acompañada de un descenso del remodelado óseo (tanto en la formación ósea como en la resorción ósea). Además, células estromales de la médula ósea de estos ratones poseen una disminución de su capacidad de formación de nódulos mineralizados y de la actividad de fosfatasa alcalina87. Paradójicamente, estos ratones deficientes en Klotho poseen un aumento del porcentaje de volumen óseo/volumen tisular en el hueso trabecular de las vértebras y de las metáfisis de los huesos largos; un efecto atribuido al aumento selectivo de actividad de la vía Wnt en ausencia de Klotho en estas localizaciones87,88.
Uno de los factores que contribuyen a la senescencia celular es la disminución de la actividad de la telomerasa en las células somáticas89. De hecho, se ha demostrado que ratones con ausencia de telomerasa presentan un descenso en la masa ósea a los tres meses del nacimiento, asociado a una reducción de formación ósea y a defectos en la proliferación y diferenciación de los osteoblastos90. Por ello, células precursoras que mantengan sus telómeros intactos y diferenciables a osteoblastos sería una estrategia adecuada para regenerar el tejido óseo en situación de osteopenia. En este sentido, se ha demostrado que células estromales que conservan sus telómeros intactos derivadas del tejido adiposo de ratones SAMP6, que presentan un déficit de osteoprogenitores, pueden ser derivadas a células osteoblásticas con la misma capacidad de mineralización y actividad de fosfatasa alcalina que las procedentes de ratones controles91.
Papel del estrés oxidativo en la osteopenia asociada al envejecimientoEl envejecimiento se explica mayoritariamente como una consecuencia del desequilibrio en los niveles de agentes oxidantes y antioxidantes, con una mayor presencia de los primeros, en lo que se denomina «estrés oxidativo». Este aumento de la oxidación con la edad conduce a la disfunción de los sistemas de regulación metabólica y al aumento del riesgo de enfermedad92,93. En condiciones normales, la mitocondria produce niveles moderados de especies reactivas de oxigeno (ROS, utilizando las siglas inglesas de reactive oxygen species). Las alteraciones de este orgánulo durante el envejecimiento, el cual se ha considerado la primera diana de las ROS1, dan lugar a una producción excesiva de ROS, que inducen daño celular y apoptosis94,95.
La disminución de la masa y la resistencia ósea con la edad y en la menopausia parece asociarse con el estrés oxidativo tanto en roedores como en humanos55,96. La generación de ROS —anión superóxido (O2−), radicales OH.− y peróxido de hidrógeno (H2O2)— por la oxidacion de ácidos grasos o en respuesta a la inflamación, disminuye la supervivencia osteoblástica55. Las ROS se asocian a la fosforilación de p66Shc, una proteína responsable de amplificar la respuesta oxidativa y de la inducción de apoptosis osteoblástica y osteocítica55,97.
La activación de factores de transcripcion de la familia FoxO (del inglés forkhead box protein) tiene lugar como mecanismo de defensa contra el estrés oxidativo6. Este mecanismo implica la interacción de FoxOs con β-catenina para inducir la expresión de genes como el de la catalasa, evitando así la interacción de β-catenina con factores de transcripción de linfocitos T (TCF), necesaria para estimular la diferenciación osteoblástica55. Con la edad, se ha demostrado una disminución de la transcripción de genes diana de β-catenina/TCF, un aumento de los modulados por FoxOs en relación con el estrés oxidativo y la disminución de la formación ósea en ratones57. El papel crítico de FoxOs en el metabolismo óseo viene avalado por estudios en ratones con manipulación genética de su expresión. Así, la eliminación génica de FoxO 1, 3 y 4 aumenta el estrés oxidativo en el hueso e induce una pérdida de masa ósea (a nivel trabecular y cortical) debida a la mayor apoptosis osteoblástica y a la disminución de formación ósea en estos ratones98.
Por otra parte, la formación de lípidos oxidados dependiente de lipoxigenasas juega también un papel importante en la osteopenia del envejecimiento. Se ha comprobado que la expresión de varias lipoxigenasas (como Alox12 y Alox15), así como el 4-hidroxinonenal (un producto de peroxidación lipídica), aumentan en el hueso de ratón durante el envejecimiento. Estos cambios se asocian al aumento del receptor activador de la proliferación de peroxisomas (PPAR) —que aumenta la capacidad adipogénica en la médula ósea— y la osteopenia99.
Diversos estudios sugieren la posible interacción del estrés oxidativo con factores osteogénicos. Así, se ha demostrado que los productos de oxidación lipídica inhiben la acción de algunos de estos factores100,101. Además, recientemente, se ha demostrado en ratones un aumento de la acción anabólica ósea de la PTH con la edad, en relación con sus propiedades antioxidantes102. Por otra parte, las ROS actúan como moléculas de señalización para inducir la transcripción del VEGF por la angiotensina II, un factor proinflamatorio103. A este respecto, nuestro grupo ha demostrado que la PTHrP ejerce acciones pro-inflamatorias en varios tipos celulares incluyendo los osteoblastos, en parte a través de la inducción de genes sensibles al sistema redox, como la proteína quimiotáctica de monocitos-1 y la interleuquina-6104–106. Más recientemente, hemos encontrado que la PTHrP es capaz de contrarrestar el estrés oxidativo inducido por el H2O2 en células osteoprogenitoras de ratón en relación con su acción osteogénica107.
Todos estos datos en conjunto apoyan la hipótesis de que el estrés oxidativo es primordial para explicar las alteraciones óseas asociadas al envejecimiento. De este modo, los mecanismos relacionados con dicho estrés podrían constituir posibles dianas moleculares para el diseño de agentes terapéuticos en el contexto de la pérdida de masa ósea asociada a la edad.
ConclusionesLos datos actuales sugieren que el deterioro óseo que tiene lugar al envejecer no es sólo consecuencia de alteraciones hormonales, sino que se debe en gran medida a un aumento del estrés oxidativo. Tanto la disminución de los niveles de hormonas sexuales, que a su vez contribuye a disminuir los mecanismos de defensa contra el estrés oxidativo, como el aumento de la peroxidación lipídica y de los glucocorticoides endógenos (ambos hechos, en parte debidos también al estrés oxidativo) son procesos que promueven el deterioro óseo. Estos factores unidos a la pérdida de función osteoblástica y a la falta de respuesta a factores osteogénicos contribuyen a la aparición de la osteopenia/osteoporosis al avanzar la edad (fig. 1). Por todo ello, es de suma importancia el desarrollo de modelos animales que permitan estudiar de modo pleiotrópico estos mecanismos y su impacto relativo en la osteoporosis involutiva. Estos modelos serán de gran utilidad para diseñar terapias idóneas para promover la formación y la regeneración ósea en las fracturas en pacientes de edad avanzada.
 y factor de crecimiento similar a insulina tipo-1 (IGF-1). El envejecimiento produce un aumento de estrés oxidativo que se traduce en unos mayores niveles de ROS. Esto provoca que el factor de transcripción β-catenina se una a FoxO para transcribir genes relacionados con la respuesta oxidativa en vez de genes osteogénicos. La aparición de ROS induce la fosforilación de p66Shc, lo que conlleva a un aumento de la apoptosis de osteoblastos y osteocitos. Todo ello, en conjunto contribuye al deterioro de la masa y de la calidad ósea. Imagen: microtomografía computarizada (μCT) de la tibia de ratones CD-1 hembras de 6 meses (izquierda) y de 18 meses (derecha) de edad, realizada por la Dra. Francisca Mulero en la Unidad de Imagen del Centro Nacional de Investigaciones Oncológicas (CNIO, Madrid).")
Mecanismos propuestos por los cuales el envejecimiento produce deterioro óseo. Al envejecer, tiene lugar una disminución de la función osteoblástica, de la expresión del receptor del calcitriol y de la respuesta por parte de los osteoblastos a la acción de la PTH, factor de crecimiento transformante β (TGF-β) y factor de crecimiento similar a insulina tipo-1 (IGF-1). El envejecimiento produce un aumento de estrés oxidativo que se traduce en unos mayores niveles de ROS. Esto provoca que el factor de transcripción β-catenina se una a FoxO para transcribir genes relacionados con la respuesta oxidativa en vez de genes osteogénicos. La aparición de ROS induce la fosforilación de p66Shc, lo que conlleva a un aumento de la apoptosis de osteoblastos y osteocitos. Todo ello, en conjunto contribuye al deterioro de la masa y de la calidad ósea. Imagen: microtomografía computarizada (μCT) de la tibia de ratones CD-1 hembras de 6 meses (izquierda) y de 18 meses (derecha) de edad, realizada por la Dra. Francisca Mulero en la Unidad de Imagen del Centro Nacional de Investigaciones Oncológicas (CNIO, Madrid).
Los Dres. Sergio Portal-Núñez y Daniel Lozano poseen contratos post-doctorales del Instituto de Salud Carlos III (RETICEF RD006/0013/1002) y de la Comunidad Autónoma de Madrid (S-2009/MAT-1472), respectivamente. Los trabajos de nuestro grupo citados en esta revisión han sido llevados a cabo con ayudas del Ministerio de Ciencia e Innovación, del Instituto de Salud Carlos III y de la Comunidad Autónoma de Madrid.
Red Temática de Investigación Cooperativa en Envejecimiento y Fragilidad (RETICEF; RD006/0013/1002) del Instituto de Salud Carlos III, Madrid.
Red Temática de Investigación Cooperativa en Envejecimiento y Fragilidad (RETICEF; RD006/0013/0003) del Instituto de Salud Carlos III, Madrid.
recomendados

www.publicationethics.org.