










La enfermedad de Charcot-Marie-Tooth es la neuropatía hereditaria más frecuente con una prevalencia en España de 28,2 casos/100.000 habitantes. Se trata de un síndrome polineuropático sensitivo-motor, desmielinizante o axonal, que puede transmitirse con herencia autosómica dominante, autosómica recesiva, o ligada al cromosoma X. Pese a su semiología estereotipada, es un síndrome genéticamente complejo, dado que se han localizado 36 loci con una treintena de genes mutantes clonados. Analizamos los mecanismos patogénicos de estas mutaciones génicas. Abordamos la fisiopatología del pie cavo, que es manifestación cardinal de la enfermedad. En estadios clínicos iniciales, el pie cavo probablemente sea desencadenado por una desnervación selectiva de la musculatura intrínseca del pie, que causa un desequilibrio entre sus músculos intrínsecos y extrínsecos con dedos en garra, retracción de la fascia plantar, elevación del arco plantar, y acortamiento del tendón de Aquiles. Revisamos el diagnóstico y tratamiento de la enfermedad.
Charcot-Marie-Tooth disease is the most frequent inherited neuropathy with a prevalence ratio in Spain of 28.2 cases/100,000 inhabitants. It is a sensory-motor polyneuropathic syndrome, either demyelinating or axonal, which might be transmitted with autosomal dominant, autosomal recessive or X-linked pattern. Despite presenting with a stereotyped semiology, this a genetically complex syndrome comprising 36 localized loci with 30 cloned mutated genes. Here we briefly review the pathogenic mechanisms of these gene mutations. We address the pathophysiology of pes cavus, which is a cardinal manifestation of the disease. In the early clinical stages, forefoot pes cavus is most probably due to selective denervation of foot musculature, and particularly of the lumbricals, which causes an imbalance between intrinsic and extrinsic foot muscles leading to toe clawing, retraction of plantar fascia, approximation of the pillars of the longitudinal arch, and shortening of the Achilles tendon. We review the disease diagnosis and treatment.
La enfermedad de Charcot-Marie-Tooth (CMT) es la neuropatía hereditaria más frecuente con una prevalencia en España de 28,2 casos por 100.000 habitantes1. En 1886, la enfermedad fue independientemente descrita en Francia por Charcot y Marie2, y en Inglaterra por Tooth3. Pocos años después, Dejerine y Sottas4 reportaron una variante más grave y precoz de la enfermedad. Estas descripciones originales son piezas maestras de la semiología clínica, motivo por el cual las resumimos a continuación.
Tooth describió el caso de cinco pacientes de CMT cuyas edades comprendían entre 7 y 49 años y el inicio sintomático entre 6 y 35 años3. Tres pacientes eran esporádicos, un paciente tenía un hermano afecto, y en el paciente restante la madre estaba afecta. La semiología fundamental era una atrofia muscular progresiva que se iniciaba en la musculatura de las piernas, a menudo en los músculos peroneales, aunque también afectaba a los músculos tibiales anteriores, extensores largos de los dedos o gemelos; tal distribución topográfica de la amiotrofia condujo al autor a proponer que la enfermedad fuera designada como tipo peroneal de la atrofia muscular progresiva. La amiotrofia de las piernas fue asimétrica en dos pacientes (fig. 1A), un hallazgo que ocurre en torno al 20% de los casos de CMT6. La atrofia de las manos y arreflexia cuadricipital se reseña en dos casos. Un paciente tenía pies cavos. La sensibilidad estaba preservada.
 Copia a plumilla de la Figura 3 de Tooth3 demostrativa de una atrofia muscular peroneal asimétrica que el autor describe del siguiente modo: Right leg. Peronei, tibialis anticus, and extensor longus digitorum are very flabby, but no so far gone as calf muscles. (B, C) Copia de las figuras 2 y 3 de Dejerine y Sottas4 correspondientes al caso Hug (Fanny). (B) Nótese la atrofia masiva de los músculos de ambas piernas, y la acusada deformidad de ambos pies en cavo-varo con garra de los dedos. (C) Atrofia de la musculatura de la mano, descrita por los autores como Mains simiennes. Atrophie des thénars et des interosseux sans griffe cubitale. Nótese también el aplanamiento de los músculos del antebrazo. Tomado de Berciano et al5 con autorización.")
(A) Copia a plumilla de la Figura 3 de Tooth3 demostrativa de una atrofia muscular peroneal asimétrica que el autor describe del siguiente modo: Right leg. Peronei, tibialis anticus, and extensor longus digitorum are very flabby, but no so far gone as calf muscles. (B, C) Copia de las figuras 2 y 3 de Dejerine y Sottas4 correspondientes al caso Hug (Fanny). (B) Nótese la atrofia masiva de los músculos de ambas piernas, y la acusada deformidad de ambos pies en cavo-varo con garra de los dedos. (C) Atrofia de la musculatura de la mano, descrita por los autores como Mains simiennes. Atrophie des thénars et des interosseux sans griffe cubitale. Nótese también el aplanamiento de los músculos del antebrazo. Tomado de Berciano et al5 con autorización.
La serie de Charcot y Marie estaba constituida por cinco pacientes con edades comprendidas entre los 7 y 25 años, e inicio sintomático entre los 3 y 15 años. Tres de ellos eran esporádicos y los dos restantes hermanos (casos 2 y 3)2. Los autores ilustraron el artículo con seis excelentes fotografías. Aunque el cuadro clínico era similar al reportado por Tooth, las fotografías del Caso 2, tomadas a los 11 años, ilustran una acusada atrofia de manos y piernas con deformidad en valgo del pie izquierdo, y deformidad en varo del derecho. A diferencia de la serie de Tooth, se detectó arreflexia de extremidades inferiores en todos los casos, si bien solo hubo hipoestesia en uno de ellos.
Dejerine y Sottas4 reportaron el caso de un hermano y hermana con grave semiología polineuropática sensitivo-motora de inicio infantil (fig. 1B y C), engrosamiento palpable de los nervios de las extremidades, cifoescoliosis y pupila de Argyll-Robertson, cuyos padres no estaban afectos, lo cual sugiere una transmisión autosómica recesiva (AR). El estudio necrópsico de ambos pacientes demostró una neuropatía hipertrófica4,7.
En síntesis, estas descripciones originales resumen muchas de las características de CMT, a saber: 1) presentación esporádica o familiar; 2) en casos familiares, transmisión autosómica dominante (AD) o AR; y 3) semiología variable de unos pacientes a otros, aunque en general con predominio de la motora sobre la sensitiva.
En las décadas siguientes, estudios clínicos y neurofisiológicos permitieron delimitar que CMT puede transmitirse con herencia AD, AR o ligada al cromosoma X, y que en función del rango de conducción nerviosa hay formas desmielinizantes (velocidad de conducción motora [VCM] de nervio mediano, < 38 m/s), axonales (VCM motora de nervio mediano, > 38 m/s) e intermedias (VCM motora de nervio mediano, 30-40 m/s)8,10. En buena correlación con las descripciones neurofisiológicas, estudios histológicos del sistema nervioso periférico (SNP) demostraron un patrón dual, ya desmielinizante o axonal (fig. 2). De este modo, en la década de los 70, Dyck13 propuso una sencilla clasificación, unánimemente aceptada, que incluye los siguientes tipos: 1) tipo I (CMT1, hipertrófico o desmielinizante) con herencia AD o AR; 2) tipo II (CMT2, neuronal o axonal) con herencia AD o AR; 3) tipo III (usualmente con herencia AR) reservado para la enfermedad de Dejerine–Sottas o pacientes con formas graves de CMT hipomielinizante; 4) formas ligadas al cromosoma X; y 5) formas complejas (e.g., con atrofia óptica, sordera o degeneración pigmentaria de la retina). Aunque en la literatura la enfermedad ha sido también designada como neuropatía motora y sensitiva hereditaria (HMSN), en la actualidad se prefiere usar el acrónimo CMT. Merece la pena reseñar que el único signo clínico diferencial entre CMT1 y CMT2 es la presencia de engrosamiento, visible o palpable, de troncos nerviosos en CMT1 (ver más adelante).
 Sección semifina de nervio sural de un paciente de CMT1A. Hay una acusada pérdida de fibras mielínicas, varias imágenes de proliferación de las células de Schwann con formación de bulbos de cebolla (flecha), y una fibra con remielinizada (punta de flecha). (B) Sección semifina de nervio ciático poplíteo externo de un paciente fallecido de CMT2G11,12. Hay marcada pérdida de fibras mielínicas gruesas y racimos de fibras en regeneración (flecha); nótese la ausencia de fenómenos hipertróficos.")
(A) Sección semifina de nervio sural de un paciente de CMT1A. Hay una acusada pérdida de fibras mielínicas, varias imágenes de proliferación de las células de Schwann con formación de bulbos de cebolla (flecha), y una fibra con remielinizada (punta de flecha). (B) Sección semifina de nervio ciático poplíteo externo de un paciente fallecido de CMT2G11,12. Hay marcada pérdida de fibras mielínicas gruesas y racimos de fibras en regeneración (flecha); nótese la ausencia de fenómenos hipertróficos.
Antes de empezar este apartado, conviene recordar que CMT tiene una relación nosológica muy estrecha con otras dos formas de neuropatía hereditaria: neuronopatía motora hereditaria distal (dHMN en el acrónimo anglosajón que usaremos aquí por ser el que figura en OMIM y PubMed) y neuropatía sensitiva y autonómica hereditaria (HSAN). Entre estos tres síndromes hay no solo solapamiento fenotípico, sino que se da el fenómeno de heterogeneidad alélica (idéntico fenotipo originado por diferentes mutaciones en el mismo gen y locus cromosómico) y de heterogeneidad de locus (mutaciones producidas en genes que se encuentran en diferentes loci cromosómicos dando lugar al mismo fenotipo). En aras de brevedad, sólo nos ocuparemos de CMT haciendo puntual referencia a dHMN y HSAN allá donde convenga.
Con la irrupción de la genética molecular hace dos décadas, la nosología de CMT ha estado en permanente cambio. Mediante análisis de ligamiento genético se han localizado 36 loci con 30 genes clonados (para recientes revisiones ver referencias9,10,14–16. En HMN/HSAN se han descrito 12 loci adicionales con 9 genes clonados. Se estima que, en su conjunto, todavía queda por descubrir la base molecular de un tercio de los casos de CMT, lo cual es un reto que quizás se facilite con las nuevas técnicas de whole-genome sequencing17,18. Estos genes y sus respectivas proteínas constituyen un microarray de moléculas que son necesarias para el normal funcionamiento del SNP. No deja de ser una ironía que CMT, pese a la aparente simplicidad de su repertorio semiológico, haya resultado ser uno de los cuadros neurológicos genéticamente más complejos.
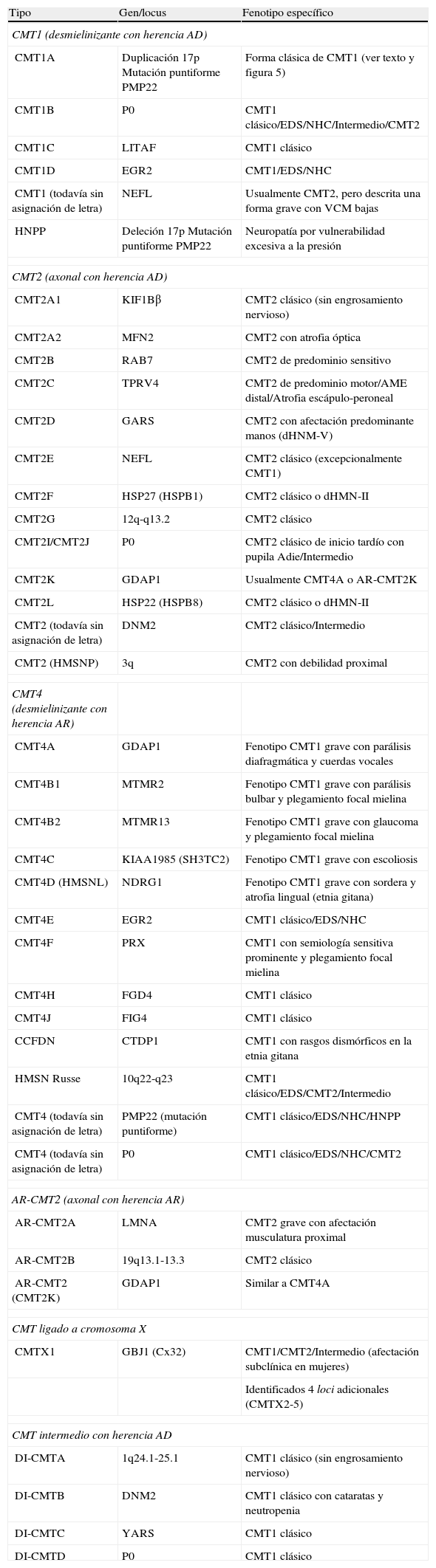
Adaptada de las referencias 10, 14 y 16, en la tabla 1 se recoge una clasificación clínico-genética actualizada de CMT, que es provisional dado que no hay un criterio unánime en el uso de sus tipos y subtipos. Hay acuerdo universal en aceptar CMT1 como cabecera para los fenotipos desmielinizante con herencia AD. Bajo CMT2, hay autores que incluyen formas axonales con herencia AD o AR, mientras que otros sólo incluyen las formas AD, creando el acrónimo AR-CMT2 para las formas axonales con transmisión AR; nosotros hemos seguido este criterio. El acrónimo CMT3 en la clasificación de Dyck13, aplicado para síndromes similares al descrito por Dejerine y Sottas4, desaparece siendo substituido por CMT4 donde se engloban todos los síndromes desmielinizantes con herencia AR. Se introduce, en fin, el acrónimo DI-CMT para las formas intermedias con transmisión AD.
Clasificación clínico-genética de CMT.
| Tipo | Gen/locus | Fenotipo específico |
| CMT1 (desmielinizante con herencia AD) | ||
| CMT1A | Duplicación 17p Mutación puntiforme PMP22 | Forma clásica de CMT1 (ver texto y figura 5) |
| CMT1B | P0 | CMT1 clásico/EDS/NHC/Intermedio/CMT2 |
| CMT1C | LITAF | CMT1 clásico |
| CMT1D | EGR2 | CMT1/EDS/NHC |
| CMT1 (todavía sin asignación de letra) | NEFL | Usualmente CMT2, pero descrita una forma grave con VCM bajas |
| HNPP | Deleción 17p Mutación puntiforme PMP22 | Neuropatía por vulnerabilidad excesiva a la presión |
| CMT2 (axonal con herencia AD) | ||
| CMT2A1 | KIF1Bβ | CMT2 clásico (sin engrosamiento nervioso) |
| CMT2A2 | MFN2 | CMT2 con atrofia óptica |
| CMT2B | RAB7 | CMT2 de predominio sensitivo |
| CMT2C | TPRV4 | CMT2 de predominio motor/AME distal/Atrofia escápulo-peroneal |
| CMT2D | GARS | CMT2 con afectación predominante manos (dHNM-V) |
| CMT2E | NEFL | CMT2 clásico (excepcionalmente CMT1) |
| CMT2F | HSP27 (HSPB1) | CMT2 clásico o dHMN-II |
| CMT2G | 12q-q13.2 | CMT2 clásico |
| CMT2I/CMT2J | P0 | CMT2 clásico de inicio tardío con pupila Adie/Intermedio |
| CMT2K | GDAP1 | Usualmente CMT4A o AR-CMT2K |
| CMT2L | HSP22 (HSPB8) | CMT2 clásico o dHMN-II |
| CMT2 (todavía sin asignación de letra) | DNM2 | CMT2 clásico/Intermedio |
| CMT2 (HMSNP) | 3q | CMT2 con debilidad proximal |
| CMT4 (desmielinizante con herencia AR) | ||
| CMT4A | GDAP1 | Fenotipo CMT1 grave con parálisis diafragmática y cuerdas vocales |
| CMT4B1 | MTMR2 | Fenotipo CMT1 grave con parálisis bulbar y plegamiento focal mielina |
| CMT4B2 | MTMR13 | Fenotipo CMT1 grave con glaucoma y plegamiento focal mielina |
| CMT4C | KIAA1985 (SH3TC2) | Fenotipo CMT1 grave con escoliosis |
| CMT4D (HMSNL) | NDRG1 | Fenotipo CMT1 grave con sordera y atrofia lingual (etnia gitana) |
| CMT4E | EGR2 | CMT1 clásico/EDS/NHC |
| CMT4F | PRX | CMT1 con semiología sensitiva prominente y plegamiento focal mielina |
| CMT4H | FGD4 | CMT1 clásico |
| CMT4J | FIG4 | CMT1 clásico |
| CCFDN | CTDP1 | CMT1 con rasgos dismórficos en la etnia gitana |
| HMSN Russe | 10q22-q23 | CMT1 clásico/EDS/CMT2/Intermedio |
| CMT4 (todavía sin asignación de letra) | PMP22 (mutación puntiforme) | CMT1 clásico/EDS/NHC/HNPP |
| CMT4 (todavía sin asignación de letra) | P0 | CMT1 clásico/EDS/NHC/CMT2 |
| AR-CMT2 (axonal con herencia AR) | ||
| AR-CMT2A | LMNA | CMT2 grave con afectación musculatura proximal |
| AR-CMT2B | 19q13.1-13.3 | CMT2 clásico |
| AR-CMT2 (CMT2K) | GDAP1 | Similar a CMT4A |
| CMT ligado a cromosoma X | ||
| CMTX1 | GBJ1 (Cx32) | CMT1/CMT2/Intermedio (afectación subclínica en mujeres) |
| Identificados 4 loci adicionales (CMTX2-5) | ||
| CMT intermedio con herencia AD | ||
| DI-CMTA | 1q24.1-25.1 | CMT1 clásico (sin engrosamiento nervioso) |
| DI-CMTB | DNM2 | CMT1 clásico con cataratas y neutropenia |
| DI-CMTC | YARS | CMT1 clásico |
| DI-CMTD | P0 | CMT1 clásico |
AME=atrofia muscular espinal; CMT=enfermedad de Charcot-Marie-Tooth; CTDP1=CTD phosphatase subunit 1; DNM=dynamin 2; EDS=enfermedad de Dejerine-Sottas; EGR2=early growth response 2; FDG4=RhoGEF; FIG4=Ptdlns(3,5)P25-phosphatase; GARS=glycyl tRNA synthetase; GBJ1=gap junction protein beta 1; GDAP1=ganglioside induced differentation associated protein 1; HMSNL=hereditary motor and sensory neuropathy Lom; HNPP=hereditary susceptibility pressure palsy; HSP22=heat shock 22 kDa protein; HSP27=heat shock 27 kDa protein; KIF1Bβ=kinesin family member 1-Bβ; LITAF=lipopolysaccharide induced tumour necrosis factor; LMNA=lamin A/C; MFN2=mitofusin 2; MTMR2=myotubularin related protein 2; MTMR13=myotubularin related protein 13; NDRG1=N-myc downstream regulated gene; NEFL=neurofilament light polypeptide 68 kDa; NHC=neuropatía hipomielinizante congénita; PMP22=peripheral myelin protein 22; P0=myelin protein zero; PRX=periaxin; RAB7=RAB7, member RAS encogen family; SH3TC2=SH3 domain and tetratricopeptide repeats; TRPV4=transient receptor potencial vallinoid 4; YARS=tyrosyl tRNA synthetase.
La figura 3 ilustra la localización de las proteínas mutadas, que era la predecible para aquellos componentes conocidos del SNP, tales como las proteínas PMP22 y P0 de la mielina compacta. En otras situaciones, sin embargo, el descubrimiento de la proteína mutada patogénica resultó ser inesperada, como por ejemplo lo ilustra el caso de GDAP1 cuya función en el SNP se desconocía hasta la identificación de CMT4A.
, HSN2 (hereditary sensory neuropathy type 2), NTRK1 (neurotrophic tyrosine kinase receptor type 1), IKBKAP (inhibitor of kappa light poypeptide gene enhancer in B-cells) y NGF1 (nerve growth factor beta polypeptide) están involucradas en la etiopatogenia de la neuropatías sensitivas y autonómicas hereditarias, no revisadas en este trabajo (ver texto).")
Dibujo esquemático de una fibra nerviosa mielinizada adaptado de Niemann et al14. Las proteínas mutadas, causales de CMT, HMN o HSAN, identificadas hasta 2006 figuran en negro, mientras que las descritas con posterioridad aparecen en rojo. Se mantienen las designaciones y acrónimos anglosajones porque son los que figuran en PubMed y OMIM. El significado de los acrónimos se recoge en el pie de la tabla 1. Nótese que mutaciones de SPTLC1 (serine palmitoyltransferase long chaín base subunit 1), HSN2 (hereditary sensory neuropathy type 2), NTRK1 (neurotrophic tyrosine kinase receptor type 1), IKBKAP (inhibitor of kappa light poypeptide gene enhancer in B-cells) y NGF1 (nerve growth factor beta polypeptide) están involucradas en la etiopatogenia de la neuropatías sensitivas y autonómicas hereditarias, no revisadas en este trabajo (ver texto).
Desde un punto de vista didáctico, los mecanismos etiopatogénicos de las proteínas mutadas se resumen del siguiente modo14: 1) por alteración del desarrollo y mantenimiento de la mielina; 2) por alteración de la biosíntesis y degradación de proteínas; 3) por alteración de la endocitosis y dinámica de membranas incluyendo la mitocondrial; 4) por alteración del citoesqueleto axonal; 5) seipinopatías; y 6) canalopatías por mutación de TRPV4. Revisaremos sucintamente estos seis apartados.
En las formas de CMT1/CMT4 por mutación de ciertos componentes de la mielina, se asume que el defecto de la célula de Schwann causa des/dismielinización con axonopatía axonal secundaria, que a la postre es responsable de la semiología clínica. El síndrome más frecuente en este apartado es CMT1A, que representa el 70% de todos los casos de CMT1 y es usualmente causado por una trisomía alélica de 17p11.2 de 1,5 Mb que contiene el gen de la proteína PMP22. Tal trisomía origina un exceso de dosis génica, lo cual implica una sobreproducción de PMP22 y su acumulación en la célula de Schwann induciendo estrés de su retículo endoplásmico, que resulta en muerte celular programada. La deleción actúa reduciendo la expresión de PMP22, lo cual origina una mielina inestable que se manifiesta con un síndrome de vulnerabilidad excesiva a la presión (HNPP en el acrónimo anglosajón; ver tabla 1). En un pequeño porcentaje de casos duplicación/deleción pueden ocurrir como un fenómeno de novo. Las mutaciones puntiformes del gen de PMP22 son excepcionales, y causan fenotipos graves, ya AD (probablemente por un mecanismo de ganancia de función) o AR (pérdida de función por fallo en la síntesis de PMP22). La proteína P0 es cuantitativamente la más abundante de la mielina compacta, y elemento esencial para su compactación. En un 10% de los casos, CMT es causado por mutaciones puntiformes de P0 que resultan, ya en un fenotipo desmielinizante de inicio precoz AD (CMT1B) y excepcionalmente AR, o bien en fenotipos axonales de inicio tardío (CMT2I y CMT2J). Así, pues, la patología molecular de PMP22/P0ha desvelado que sus mutaciones pueden heredarse a través de transmisión tanto AD como AR, y que en el caso de P0 sus mutaciones causan tanto un fenotipo desmielinizante como axonal, lo cual no hace sino subrayar que en el SNP el diálogo entre las células de Schwann y los axones acompañantes es continuo14. Tales fenómenos son aplicables a mutaciones causales de CMT en otros genes (tabla 1). GBJ1 (Cx32) es una proteína tipo gap de la mielina paranodal, cuyo gen está localizado en el cromosoma X. Segunda causa en frecuencia de CMT, mutaciones puntiformes en el gen GBJ1 originan una disfunción del tránsito radial de pequeñas moléculas entre célula Schwann y axón. Probablemente por un mecanismo de haploinsuficiencia, tales mutaciones causan un fenotipo más grave en varones que en mujeres, que neurofisiológicamente puede ser desmielinizante, intermedio o axonal. Otras causas más raras de CMT1/CMT4 incluyen mutación de EGR2 (un factor transcripción involucrado en la regulación de genes de la mielina), y mutación de PRX (una proteína de anclaje del cito-esqueleto de la célula de Schwann).
La correcta composición y mantenimiento de los compartimentos membranáceos de células de Schwann y neuronas del SNP dependen de un perfecto equilibrio entre la síntesis de componentes estructurales y de señalización, y sus procesos de degradación14. Entre las mutaciones puntiformes de proteínas implicadas en los procesos de endocitosis se cuentan las siguientes (tabla 1): 1) fosfatasas (MTMR2, MTMR13 y figura 4), que causan fenotipos AR graves (CMT4B1, CMT4B2 y CMT4J) con plegamientos focales de la mielina (CMT4B1 y CMT4B2); 2) GTPasas, DNM2 con fenotipo AD que puede ser tanto intermedio (DI-CMTB) como axonal19,20, RAB7 que causa CMT2B, un fenotipo similar al de HSNA1, y FRABIN que se asocia con CMT4H; y 3) NDRG1, un gen regulador de función poco conocida, cuya mutación causa un grave síndrome (CMT4D) en sujetos de raza gitana. Por lo que respecta a componentes implicados en la síntesis, clasificación y degradación de proteínas, las mutaciones afectan a los siguientes componentes: 1) LITAF/SIMPLE, una ligasa de ubicuitina, que causa CMT1C; y 2) GARS y YARS, proteínas implicadas en la carga del ARNt con glicina y tirosina, que causan CMT2D/dHMN-V y DI-CMTC, respectivamente (tabla 1).

Las neuronas del SNP, tanto sensitivas como motoras, deben mover proteínas, vesículas y organelas, por los largos trechos axonales que van desde el soma hasta sus terminales, lo cual requiere un sistema de transporte complejo y eficiente. No puede sorprender el creciente número de formas de CMT axonal causadas por mutaciones de proteínas relacionadas con el citoesqueletocito, y el transporte de proteínas, vesículas y organelas (tabla 1). Mutaciones en la cadena ligera de los neurofilamentos (NEFL) ocasionan CMT2E, y excepcionalmente CMT1F. Las proteínas de choque térmico (HSP) son macromoléculas ubicuas que en el SNP controlan el ensamblaje de los neurofilamentos. Mutaciones de HSP27 causan CMT2F/dHMN-II, mientras que mutaciones en HSP22 se asocian con CMT2L/dHMN-II. Recientemente, en una estirpe de CMT asociada a la mutación HSP27 R127W, con 10 pacientes explorados clínica y neurofisiológicamente, había casos con fenotipo de CMT2 y otros con fenotipo de HMN, lo cual no hace sino subrayar que ambos síndromes pueden ser una y única entidad nosólogica21. Las kinesinas son una familia de proteínas motoras que median el transporte axonal anterógrado sobre los microtúbulos, mientras que las dineínas median el transporte retrógrado. Mutaciones de KIF1Bβ se asocia con CMT2A1, y mutaciones de RAB7, GTPasa que regula la función de dineínas, causa CMT2B. La morfología mitocondrial es determinada por un equilibrio entre procesos de fusión y fisión de la organela. MNF2 es una GTPasa de la pared externa de la mitocondria, donde actúa como regulador de la fusión mitocondrial. Mutaciones puntiformes de MFN2 causan CMT2A2, siendo actualmente la causa más frecuente de CMT2 (20%), con una quinta parte de los casos presentándose como mutaciones de novo. Remedando HMSN-VI, en CMT2A2 puede haber atrofia óptica, especialmente en formas graves de inicio precoz. GDAP1 es la contrapartida de MFN2 participando en procesos de fisión mitocondrial. Mutaciones homocigotas de GDAP1 causan ya CMT4A o bien AR-CMT2; excepcionalmente ciertas mutaciones en este gen causan enfermedad en estado heterocigoto (CMT2K). En España las mutaciones de GDAP1 son las más frecuentes en CMT con herencia AR; se trata de un fenotipo grave, usualmente acompañado de parálisis de las cuerdas vocales y del diafragma22. LMNA es una proteína de la membrana nuclear cuya mutación se asocia con AR-CMT2A; tiene interés señalar que mutaciones en el mismo gen pueden causar la miopatía de Emery-Dreifuss. KIAA1985/SH3TC2 es una proteína adaptadora y sus mutaciones causan un fenotipo grave (CMT4C)17 (tabla 1).
BSCL2 es un acrónimo derivado de Berardinelli-Seip Congenital Lipodystropy 2, un síndrome originalmente descrito en estirpes con lipoatrofia, resistencia a la insulina, hipertrigliciremia, retraso mental y herencia AD. BSCL2 o Seipin es una proteína glicosilada del retículo endoplásmico, cuyas mutaciones puntiformes activan la vía UPR (unfolded protein response) induciendo estrés del retículo endoplásmico y muerte celular programada23. Las seipinopatías están consideradas como un nuevo modelo de enfermedad por alteración de la conformación proteica. Mutaciones puntiformes causan un continuo de síndromes neurodegenerativos con transmisión AD, que incluyen dHMN-V, síndrome de Silver (paraparesia espástica y amiotrofia de manos), CMT2, y paraparesia espástica hereditaria; en un estimable porcentaje de casos, la mutación tiene penetrancia incmpleta24.
TRPV4 es un miembro de canales catiónicos no selectivos implicados en la detección de estímulos físicos y químicos y en múltiples funciones fisiológicas25. Mutaciones heterocigotas de TRPV4 se habían asociado con displasias óseas. Por análisis de ligamiento genético se sabía que CMT2C, la forma escápulo-peroneal de la atrofia muscular espinal (AME) y la forma congénita distal de AME podían ser síndromes alélicos (12q21-q24). Cuatro recientes estudios han demostrado que, en efecto, tales síndromes, a veces con penetrancia incompleta, se asocian a diversas mutaciones puntuales heterocigóticas en el dominio ankirina de TRPV426–29. Se desconoce el mecanismo por el que tales mutaciones causan degeneración del SNP. En todo caso, la enfermedad es un ejemplo prototípico de expresividad variable inter e intrafamiliar.
Diagnóstico de CMTEl primer paso es establecer si el paciente padece una neuropatía hereditaria. La respuesta puede ser evidente cuando la encuesta familiar demuestra que en la estirpe hay ancestros afectos, lo cual sugiere una herencia AD, o ligada al sexo (cuando no hay transmisión varón-varón). La ocurrencia de enfermedad entre hermanos y la consanguinidad paterna sugiere una herencia AR. A veces, sin embargo, la encuesta familiar es negativa, en cuyo caso hay una serie de factores que orientan a una neuropatía genética, a saber: 1) presentación en la infancia; 2) curso clínico prolongado y lentamente progresivo; 3) presencia de pie cavo (ver más abajo); y 4) a diferencia de las neuropatías adquiridas, ausencia de síntomas sensitivos positivos (parestesias o disestesias) pese a que haya clara semiología de déficit sensitivo16. Dado que a menudo los sujetos afectos tienen síntomas sutiles o incluso están asintomáticos, junto al probando es importante explorar el máximo número posible de sujetos en riesgo de la estirpe (casos secundarios). Esto posibilita detectar signos mínimos de enfermedad (e.g., pie cavo o arreflexia) en casos subclínicos y, de este modo, perfilar mejor el patrón de herencia.
El paso siguiente es el examen neurofisiológico que debe incluir la determinación de la VCM y VCS en al menos tres nervios. A la hora de interpretar el grado de lentitud de la VCM deberá tomarse en consideración la amplitud de los potenciales de acción motores compuestos (CMAP), porque una acusada caída de la amplitud del CMAP distal implica pérdida de fibras gruesas dependiente de la distancia, que puede llevar aparejada una reducción proporcional de la VCM. Para discernir entre caída de VCM por axonopatía o mielinopatía, se recomienda estudiar segmentos proximales del nervio, donde la conducción estará similarmente lentificada en casos de CMT desmielinizante y menos lentificada e incluso preservada en casos de CMT axonal. En CMT1/CMT4 la lentificación de la VCM/VCS es difusa y uniforme, y la morfología de los CMAP y el índice de latencia terminal suelen estar preservados, lo cual está en contraposición con lo que acontece en las neuropatías inflamatorias adquiridas.
Actualmente la biopsia de nervio queda reservada para casos en los que se plantean problemas de diagnóstico diferencial con otras neuropatías hereditarias (e.g., amiloidosis) o con neuropatías adquiridas.
De la treintena de genes patogénicos hasta ahora identificados sólo una decena de ellos están disponibles para fines diagnósticos en la práctica clínica. Los tests moleculares son además caros. Así, pues, la selección de las pruebas genéticas es de suma importancia y ha de basarse en los datos clínicos así como en la frecuencia de los diversos genotipos en el país o región de estudio.
En la figura 4 se recoge el algoritmo diagnóstico ante un paciente con CMT desmielinizante. Si la herencia es AD y considerando que CMT1A es la forma más frecuente de CMT1, debe procederse al análisis de la duplicación 17p; su detección es diagnóstica de CMT1A. El marco clínico usual es el de un síndrome de atrofia muscular peroneal con arreflexia generalizada, ligera hipoestesia distal en calcetín, y otros signos ilustrados en la figura 5. El inicio sintomático suele ocurrir en las dos primeras décadas de la vida, si bien no es rara la existencia de casos subclínicos8,-10,30,31. Las VCM suelen estar en torno a los 20 m/s30,31. Si no hay duplicación ni evidencia de transmisión varón-varón debe procederse al escrutinio de mutaciones en Cx32, especialmente si la velocidad de conducción está en rango intermedio. La siguiente mutación en frecuencia es la de P0, sobre todo si la VCM está en torno a los 10 m/s32. Cuando el análisis molecular es negativo, pueden buscarse mutaciones puntiformes en PMP22, LITAF/SIMPLE, NEFL y GDAP1. Si hay evidencia de transmisión varón-varón, la estrategia es idéntica, pero omitiendo el análisis de Cx32. Las formas AR (CMT4) son especialmente frecuentes en países o regiones con fuerte endogamia. Como en la descripción original de Dejerine y Sottas4, se trata de formas graves de inicio infantil o congénito. Las velocidades de conducción nerviosa, cuando se obtienen, suelen estar por debajo de los 10 m/s. Por tratarse de un grupo sindrómico genéticamente complejo, es esencial guiarse por los datos de la epidemiología genética. De este modo, en sujetos de raza gitana, el primer paso es descartar mutaciones en NDRG4, mientras que en otras etnias de la población española han de considerarse primero P0, GDAP1 y SH3TC2, y después las restantes ocho mutaciones referidas en el algoritmo (fig. 4). En las formas ligadas al cromosoma X y en casos esporádicos se procede como indica el algoritmo.
 Engrosamiento del nervio auricular en una paciente de 8 años. (B, C) Atrofia muscular peroneal en un paciente de 17 años; nótese la presencia de dedos en garra, la inversión calcánea con adducción de antepie, y la desviación en varo del tobillo. (D-G) Fotografías de aproximación que ilustran el pie cavo en visión lateral y plantar, la garra de los dedos de los pies, y la atrofia del pedio. (H, I) Amiotrofia de las manos en una paciente de 75 años; este signo suele ser propio de estadios avanzados de la enfermedad.")
Composición de los signos clínicos en CMT1A en enfermos estudiados por los autores. (A) Engrosamiento del nervio auricular en una paciente de 8 años. (B, C) Atrofia muscular peroneal en un paciente de 17 años; nótese la presencia de dedos en garra, la inversión calcánea con adducción de antepie, y la desviación en varo del tobillo. (D-G) Fotografías de aproximación que ilustran el pie cavo en visión lateral y plantar, la garra de los dedos de los pies, y la atrofia del pedio. (H, I) Amiotrofia de las manos en una paciente de 75 años; este signo suele ser propio de estadios avanzados de la enfermedad.
En la figura 6 se recoge el algoritmo diagnóstico en un paciente con CMT axonal. Si la herencia es AD y no hay evidencia de transmisión varón-varón, el estudio molecular empieza con Cx32, para seguir con las mutaciones de MFN2, P0 y DNM2, y después otras nueve mutaciones génicas menos frecuentes (ver algoritmo). Si hay evidencia de transmisión varón-varón, el escrutinio molecular es idéntico, pero omitiendo Cx32. En las formas con transmisión AR, la mutación de GDAP1 es la más frecuente; después cabe considerar las de LMNA y NEFL. Para casos con herencia al cromosoma X y esporádicos se procede como se indica en el algoritmo.
Pie cavo en CMT
El pie cavo es una manifestación cardinal de la enfermedad8,9,13,30,31. Como indican las figuras 1 y 5, se trata de un pie cavo y valgo del antepie (forefoot cavus), usualmente acompañado de dedos en garra y deformidad en varo del retropie33,34. La presencia de pie cavo indica que el proceso de desnervación de la musculatura del pie se inició antes de completarse su crecimiento35,36. La fisiopatología del pie cavo en CMT es una cuestión controvertida. Hay acuerdo general, que el pie cavo es causado por un desequilibrio entre músculos agonistas y antagonistas de la musculatura del pie o de la pierna. En la literatura ortopédica, hay coincidencia en señalar que los dedos en garra son la consecuencia de una paresia de la musculatura intrínseca del pie con preservación de la extrínseca34. En relación con el mecanismo del pie cavo en CMT hay dos hipótesis divergentes33-37. Según la primera hipótesis, propuesta por la mayoría de autores, la deformidad en cavo del antepie es la consecuencia de un desequilibrio entre la fuerza preservada del músculo peroneo lateral largo y una paresia del peroneo lateral corto y/o del tibial anterior. La acción descompensada del peroneo lateral largo causaría una flexión plantar excesiva del primer metatarsiano con incremento de la altura del arco plantar y desviación subastragalina en varo. La segunda hipótesis propone que el pie cavo resulta de una desnervación de la musculatura intrínseca del pie, y particularmente de los lumbricales, con relativa preservación de la musculatura extrínseca, es decir, tanto los dedos en garra como el pie cavo no serían sino la expresión de un desequilibrio en la acción la musculatura intrínseca (desnervada) y extrínseca (preservada) del pie38.
La limitación principal en la interpretación fisiopatológica del pie cavo en CMT es que se sustentaba en estudios transversales de casos sintomáticos, y sobre todo de casos índice en los que la presencia de amiotrofia y debilidad de las piernas es la regla. Faltaban estudios longitudinales clínico-neurofisiológicos de casos secundarios (subclínicos) para establecer la evolución de la semiología del pie. Nosotros abordamos esta cuestión explorando prospectiva y consecutivamente, a lo largo de dos décadas, 20 niños en riesgo de CMT1A con duplicación (madre o padre enfermos), de los cuales 12 resultaron estar afectos30,31,39,40. El período de inclusión fue el primer quinquenio de la vida (edades de los 12 pacientes comprendidas entre 0 y 4 años; media, 2 años), que es cuando se produce la maduración de la conducción nerviosa. Al finalizar el estudio las edades se movían entre 4 y 19 años (media, 8). En el período de inclusión sólo 4 de 12 pacientes tenían pie cavo; entre los 5 y 10 años, 5 de los 10 pacientes que habían alcanzado este grupo de edad tenía pie cavo; y a partir de los 11 años todos ellos (7 de 7) tenía pie cavo. Sólo se detectó atrofia muscular peroneal en este último grupo de edad (en 4 de ellos). Comprobamos también que la aparición y progresión de la amiotrofia en el músculo pedio se correlaciona no con el grado de lentitud de la VCM del nervio peroneal sino con la caída de su CMAP, es decir, con la axonopatía secundaria dependiente de distancia. Así, pues, nuestros estudios demostraban que el pie cavo aparece en la infancia o adolescencia y sin relación con la presencia de paresia tibio-peronea, lo cual reforzaba el papel fisiopatológico de la desnervación de la musculatura intrínseca del pie en el desarrollo del pie cavo38.
Posteriormente, para corroborar nuestros hallazgos clínico-neurofisiológicos, llevamos a cabo un estudio por resonancia magnética (RM) de la musculatura de piernas y pies en 11 pacientes de CMT1A, 6 con fenotipo leve que incluía pie cavo pero sin paresia de la musculatura de las piernas, y 5 con fenotipo más avanzado incluyendo pie cavo y paresia tibio-peronea41. Concordantemente con nuestros hallazgos clínico-neurofisiológicos previos, en los pacientes con fenotipo leve el examen RM demostró que la atrofia grasa quedaba restringida a la musculatura del pie, mientras que en pacientes con fenotipo moderado había una combinación de atrofía grasa masiva de la musculatura del pie y, en menor grado, atrofia de la musculatura de la pierna de predominio distal. Casi simultáneamente y en un trabajo de balance muscular sin precedentes en la literatura, Vinci et al demostraban que, en casos de CMT1A con fenotipo leve, la paresia puede quedar circunscrita a lumbricales y flexor corto del dedo gordo con total preservación de la musculatura tibio-peroneal42.
Resumiendo, inicialmente el pie cavo en CMT1A depende de la desnervación selectiva de la musculatura del pie, que causa un desequilibrio entre su musculatura intrínseca y extrínseca (fig. 7). El desequilibrio en la acción de la musculatura peroneo-tibial quizás tenga su papel fisiopatológico a medida que la enfermedad avanza, o en casos más graves con desnervación precoz de la musculatura del compartimento anterolateral de la pierna. Esta noción probablemente sea aplicable a otros síndromes de CMT diferentes de CMT1A con duplicación.
 La enfermedad se inicia con la desnervación de lumbricales y de otros músculos intrínsecos del pie. (B) La paresia de lumbricales origina dedos en garra, aplanamiento del arco plantar anterior y contractura de los músculos flexores cortos aproximando los pilares del arco longitudinal del pie. (C) Durante la marcha, antes de despegar los dedos, con la extensión de las articulaciones metatarsofalángicas, la aponeurosis plantar se enrosca alrededor de las cabezas metatarsianas (efecto cabrestante) aproximando todavía más los pilares del arco longitudinal del pie y acortando el tendón de Aquiles, lo cual limita la dorsiflexión del pie.")
Partiendo de imágenes tomadas del Sabir y Lyttle38y de los hallazgos RM de la musculatura de pie41, interpretación fisiopatológica del pie cavo en estadios precoces de CMT1A. (A) La enfermedad se inicia con la desnervación de lumbricales y de otros músculos intrínsecos del pie. (B) La paresia de lumbricales origina dedos en garra, aplanamiento del arco plantar anterior y contractura de los músculos flexores cortos aproximando los pilares del arco longitudinal del pie. (C) Durante la marcha, antes de despegar los dedos, con la extensión de las articulaciones metatarsofalángicas, la aponeurosis plantar se enrosca alrededor de las cabezas metatarsianas (efecto cabrestante) aproximando todavía más los pilares del arco longitudinal del pie y acortando el tendón de Aquiles, lo cual limita la dorsiflexión del pie.
El tratamiento de la enfermedad es multidisciplinario, lo cual involucra a pediatras, neurólogos, rehabilitadores y traumatólogos. El abordaje ortopédico de la enfermedad es analizado por Fernández-Retana y Poggio en un trabajo que se publica en este mismo número de la revista. Por nuestra parte, quisiéramos subrayar que CMT en niños es a menudo la expresión de una mera alteración de la arquitectura del pie, sin pérdida real de la fuerza muscular de la pierna; esto llama a un tratamiento fisioterápico conservador, que mitigue en lo posible el desarrollo dedos en garra y la retracción del tendón de Aquiles que limita la dorsiflexión del pie. El paciente debe ser animado a observar una vida lo más activa posible, controlando su peso y evitando el abuso alcohólico y la administración de fármacos neurotóxicos.
En 2004, Passage et al reportaban que el ácido ascórbico, un promotor de la mielinización, mejora el fenotipo en un modelo de CMT1A en el ratón que sobre-expresa PMP2243. Este hallazgo experimental condujo a la realización del 136th ENMC International Workshop on Charcot-Marie-Tooth disease type 1A, donde se concluyó que estaba justificado emprender ensayos clínicos con vitamina C en pacientes de CMT1A44. Desgraciadamente, los tres ensayos clínicos reportados hasta ahora han sido negativos45–47, lo cual atestigua que los modelos animales de CMT1A no necesariamente recapitulan el fenotipo humano. Para otras perspectivas terapéuticas, nos remitimos a la reciente revisión de Reilly y Shy16.
Nivel de evidenciaTema de actualización con nivel de evidencia V.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo ha sido financiado por CIBERNED y el Fondo de Investigación Sanitaria del ISCIII (PI07/132E).
